

学习内容
- 评估是否存在聚类的假象
- 用PCA和UMAP图来确定聚类的质量,并了解何时需要重新聚类
- 评估已知的细胞类型标志物以假设集群的细胞类型身份
目标
- 确定集群是否代表真正的细胞类型或由于生物或技术差异而产生的集群,如细胞周期S期的细胞集群、特定批次的集群或线粒体含量高的细胞。
- 使用已知的细胞类型标记基因来确定群组的身份。
探索指控参数
为了确定我们的聚类是否可能是由细胞周期阶段或线粒体表达等人为因素造成的,从视觉上探索这些指标,看是否有聚类表现出富集或与其他聚类不同,是很有用的。然而,如果观察到特定集群的富集或差异,如果可以用细胞类型来解释,可能并不令人担忧。
为了探索和可视化各种质量指标,我们将使用Seurat的多功能DimPlot()和FeaturePlot()函数。
按样本划分聚类
我们可以从探索每个样本中每个聚类的细胞分布开始:
# Extract identity and sample information from seurat object to determine the number of cells per cluster per sample
n_cells <- FetchData(seurat_integrated,
vars = c("ident", "orig.ident")) %>%
dplyr::count(ident, orig.ident) %>%
tidyr::spread(ident, n)
# View table
View(n_cells)
我们可以用UMAP可视化每个样本的每个簇的细胞。
# UMAP of cells in each cluster by sample
DimPlot(seurat_integrated,
label = TRUE,
split.by = "sample") + NoLegend()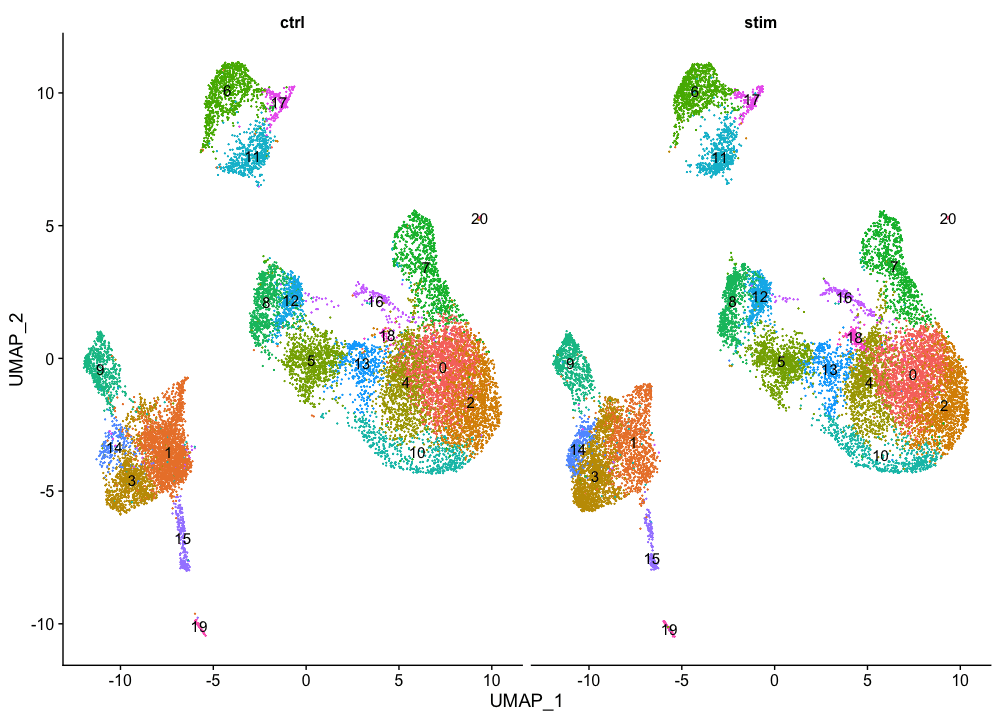
一般来说,我们希望看到大多数的细胞类型集群出现在所有的条件下;然而,根据实验,我们可能希望看到一些特定条件的细胞类型出现。这些细胞簇在不同条件下看起来非常相似,这很好,因为我们期望类似的细胞类型在对照组和刺激条件下都存在。
根据细胞周期划分聚类
接下来,我们可以探讨细胞是否按不同的细胞周期阶段聚集。当我们进行SCTransform归一化和回归无意义的变异来源时,我们并没有回归出细胞周期阶段引起的变异。如果我们的细胞集群在细胞周期表达方面表现出较大的差异,这将表明我们要重新运行SCTransform,并将S.Score和G2M.Score添加到我们的变量中进行回归,然后重新运行其余步骤。
# Explore whether clusters segregate by cell cycle phase
DimPlot(seurat_integrated,
label = TRUE,
split.by = "Phase") + NoLegend()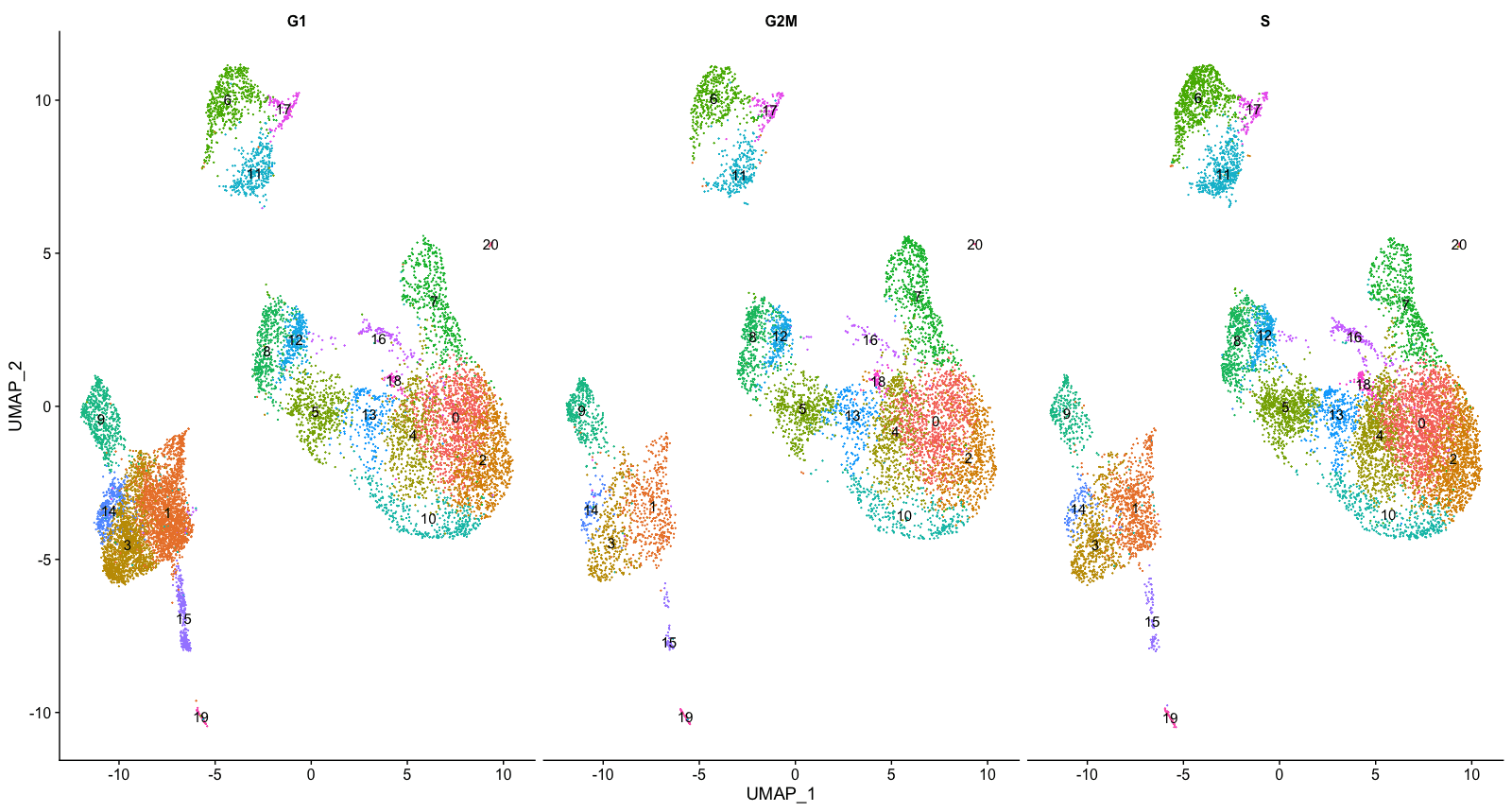
我们没有看到按细胞周期得分进行的聚类,所以我们可以继续进行质量控制。
通过各种无意义的变异来隔离集群
接下来我们将探索更多的指标,如每个细胞的UMI和基因的数量,S期和G2M期的标志物,以及通过UMAP的线粒体基因表达。观察单个S和G2M的分数可以给我们提供额外的信息来检查阶段,就像我们之前做的那样。
# Determine metrics to plot present in seurat_integrated@meta.data
metrics <- c("nUMI", "nGene", "S.Score", "G2M.Score", "mitoRatio")
FeaturePlot(seurat_integrated,
reduction = "umap",
features = metrics,
pt.size = 0.4,
order = TRUE,
min.cutoff = 'q10',
label = TRUE)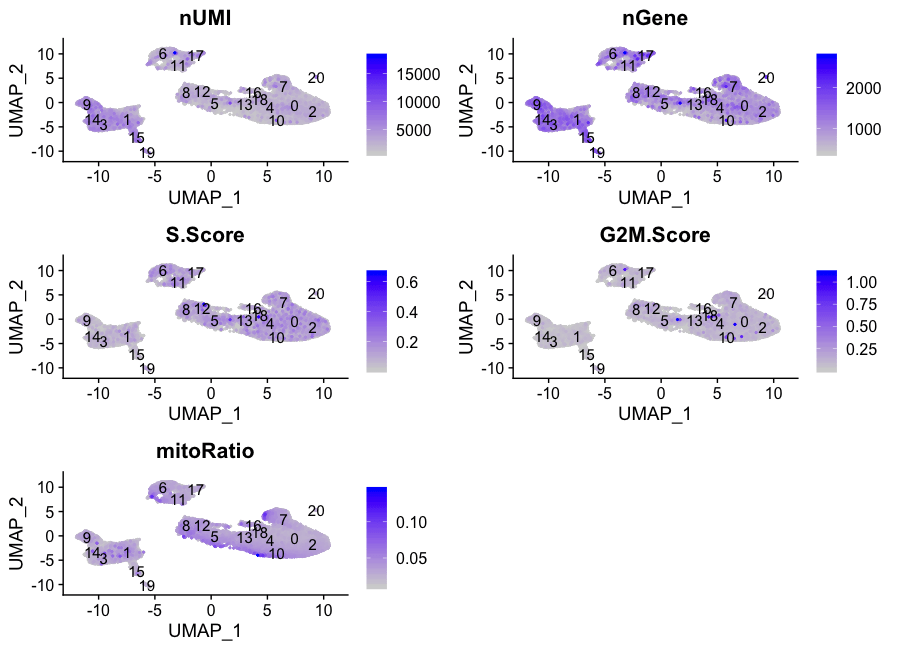
各个集群的指标似乎相对均匀,除了nUMIs和nGene在集群3、9、14和15,也许还有集群17表现出较高的数值。我们将继续关注这些群组,看看细胞类型是否可以解释这种增加。
如果我们在这个时候看到与这些指标中的任何一项相对应的差异,那么我们通常会注意到它们,然后在确定细胞类型的身份后决定是否采取任何进一步的行动。
探索驱动聚类分群的PC情况
我们还可以探索我们的集群在不同PC上的分离程度;我们希望定义的PC能很好地分离细胞类型。为了使这一信息可视化,我们需要提取细胞的UMAP坐标信息,以及它们在每个PC上的相应分数,以便通过UMAP查看。
首先,我们确定我们想从Seurat对象中提取的信息,然后,我们可以使用FetchData()函数来提取这些信息。
# Defining the information in the seurat object of interest
columns <- c(paste0("PC_", 1:16),
"ident",
"UMAP_1", "UMAP_2")
# Extracting this data from the seurat object
pc_data <- FetchData(seurat_integrated,
vars = columns)在下面的UMAP图中,细胞按照每个主成分的PC得分来标示。
让我们快速浏览一下排名前16位的PC。
# Adding cluster label to center of cluster on UMAP
umap_label <- FetchData(seurat_integrated,
vars = c("ident", "UMAP_1", "UMAP_2")) %>%
group_by(ident) %>%
summarise(x=mean(UMAP_1), y=mean(UMAP_2))
# Plotting a UMAP plot for each of the PCs
map(paste0("PC_", 1:16), function(pc){
ggplot(pc_data,
aes(UMAP_1, UMAP_2)) +
geom_point(aes_string(color=pc),
alpha = 0.7) +
scale_color_gradient(guide = FALSE,
low = "grey90",
high = "blue") +
geom_text(data=umap_label,
aes(label=ident, x, y)) +
ggtitle(pc)
}) %>%
plot_grid(plotlist = .)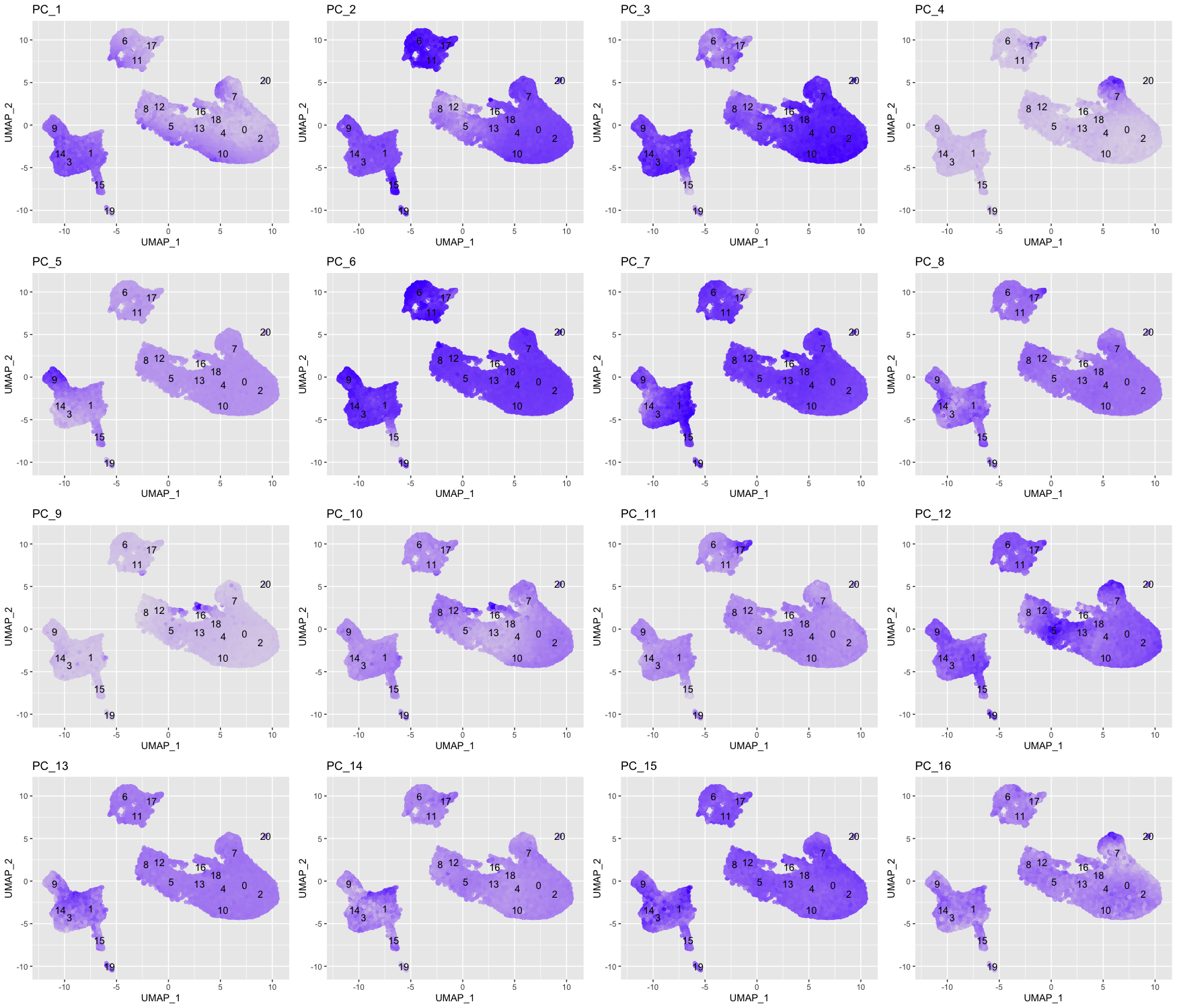
我们可以看到不同的PC是如何代表集群的。例如,驱动PC_2的基因在集群6、11和17中表现出较高的表达量(也许在15中也有点高)。我们可以回头看看我们驱动这个PC的基因,以了解细胞类型可能是什么。
# Examine PCA results
print(seurat_integrated[["pca"]], dims = 1:5, nfeatures = 5)
以CD79A和CD74基因以及HLA基因作为PC_2的阳性标记,我们可以假设第6、11和17集群对应于B细胞。这只是暗示了集群的身份可能是什么,集群的身份是通过PC的组合来确定的。
为了真正确定集群的身份以及resolution是否合适,探索少数预期的细胞类型的已知基因标记是有帮助的。
探索已知的细胞类型标记物
随着细胞的聚类,我们可以通过寻找已知的标记物来探索细胞类型的特性。标有集群的UMAP图被显示出来,然后是预期的不同细胞类型。
DimPlot(object = seurat_integrated,
reduction = "umap",
label = TRUE) + NoLegend()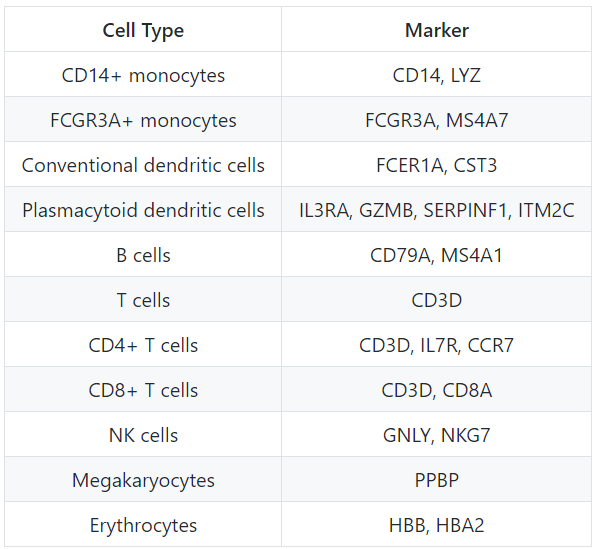
已知细胞标记物参考
Seurat的FeaturePlot()函数使我们很容易使用存储在Seurat对象中的基因ID对少数基因进行可视化。我们可以很容易地在我们的UMAP可视化的基础上探索已知基因标记的表达。让我们去确定集群的身份。为了获取所有基因的归一化表达水平,我们可以使用存储在RNA检测槽中的归一化计数数据。
SCTransform归一化只对3000个最易变的基因进行了处理,所以我们感兴趣的许多基因可能不存在于这个数据中。所以要把默认检测修改为RNA。
# Select the RNA counts slot to be the default assay
DefaultAssay(seurat_integrated) <- "RNA"
# Normalize RNA data for visualization purposes
seurat_integrated <- NormalizeData(seurat_integrated, verbose = FALSE)Assay(检测)是Seurat对象中定义的一个槽,它内部有多个槽。在一个给定的检测中,counts槽存储非归一化的原始计数,而data槽存储归一化的表达数据。因此,当我们在上面的代码中运行NormalizeData()函数时,归一化的数据将被存储在RNA检测的data槽中,而计数槽将保持不变。
根据我们感兴趣的标志物,它们可能是某一特定细胞类型的阳性或阴性标志物。我们结合少数几个标记物的综合表达应该可以大致推断出某些聚类的细胞类型。
对于这里使用的标记物,我们正在寻找阳性标记物和标记物在各群组中表达的一致性。例如,如果一个细胞类型有两个标记物,而在一个簇中只有一个标记物表达–那么我们就不能可靠地将该簇分配给该细胞类型。
CD14 +单核细胞标志物
FeaturePlot(seurat_integrated,
reduction = "umap",
features = c("CD14", "LYZ"),
order = TRUE,
min.cutoff = 'q10',
label = TRUE)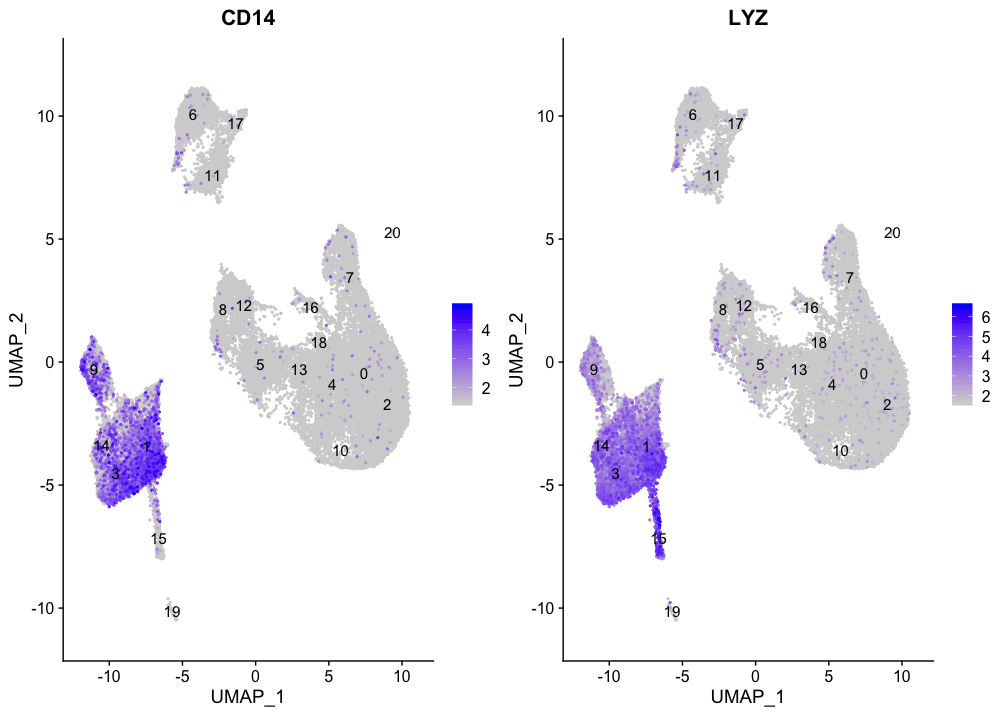
CD14+单核细胞似乎对应于1、3和14群。我们不会将集群9和15包括在内,因为它们并不高度表达这两种标记物。
FCGR3A +单核细胞标志物
FeaturePlot(seurat_integrated,
reduction = "umap",
features = c("FCGR3A", "MS4A7"),
order = TRUE,
min.cutoff = 'q10',
label = TRUE)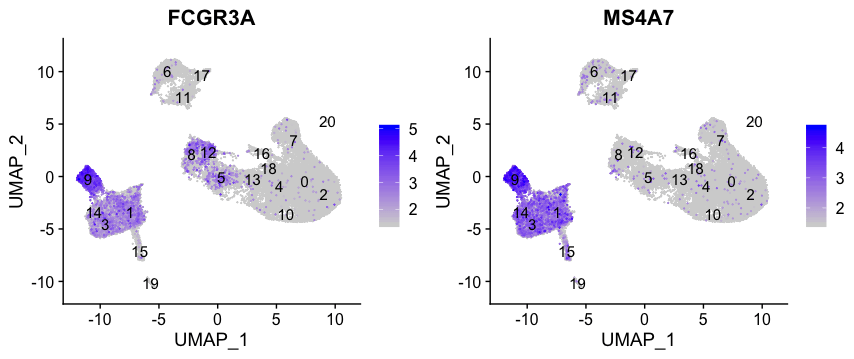
FCGR3A+单核细胞的标志物明显地突出了第9群,尽管我们在第1、3和14群中确实看到一定程度的表达。我们希望在进行标记物鉴定时看到FCGR3A+细胞的其他标记物显示出来。
巨噬细胞
FeaturePlot(seurat_integrated,
reduction = "umap",
features = c("MARCO", "ITGAM", "ADGRE1"),
order = TRUE,
min.cutoff = 'q10',
label = TRUE)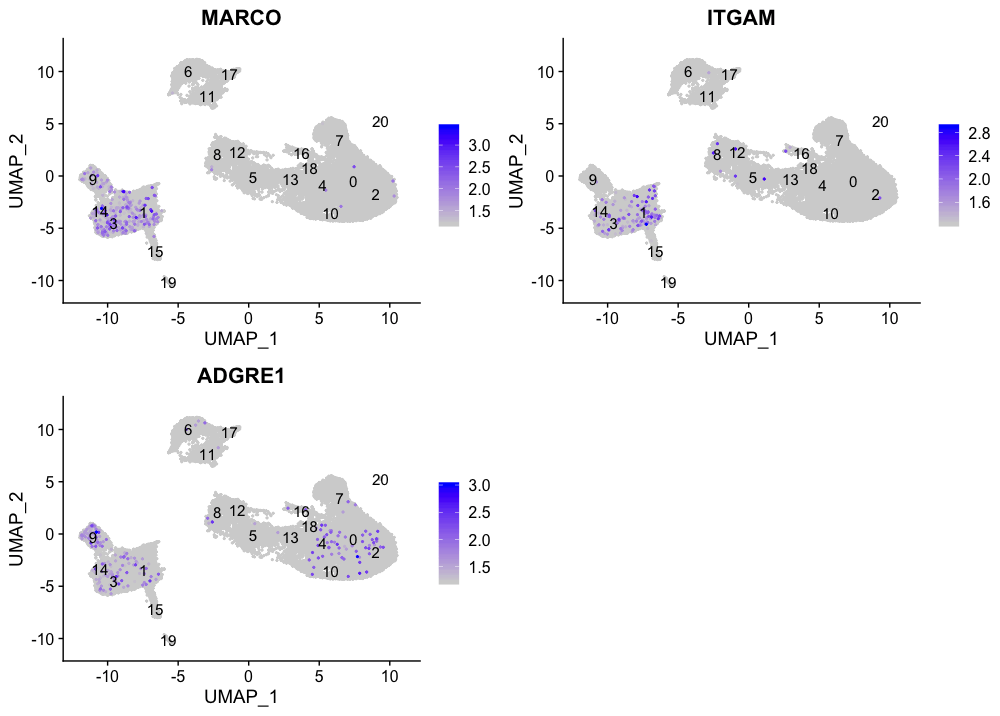
我们没有看到我们的标记物有多少重叠,所以没有集群似乎对应于巨噬细胞;也许细胞培养条件对巨噬细胞有负面选择(更高的粘附性)。
常规树突状细胞
FeaturePlot(seurat_integrated,
reduction = "umap",
features = c("FCER1A", "CST3"),
order = TRUE,
min.cutoff = 'q10',
label = TRUE)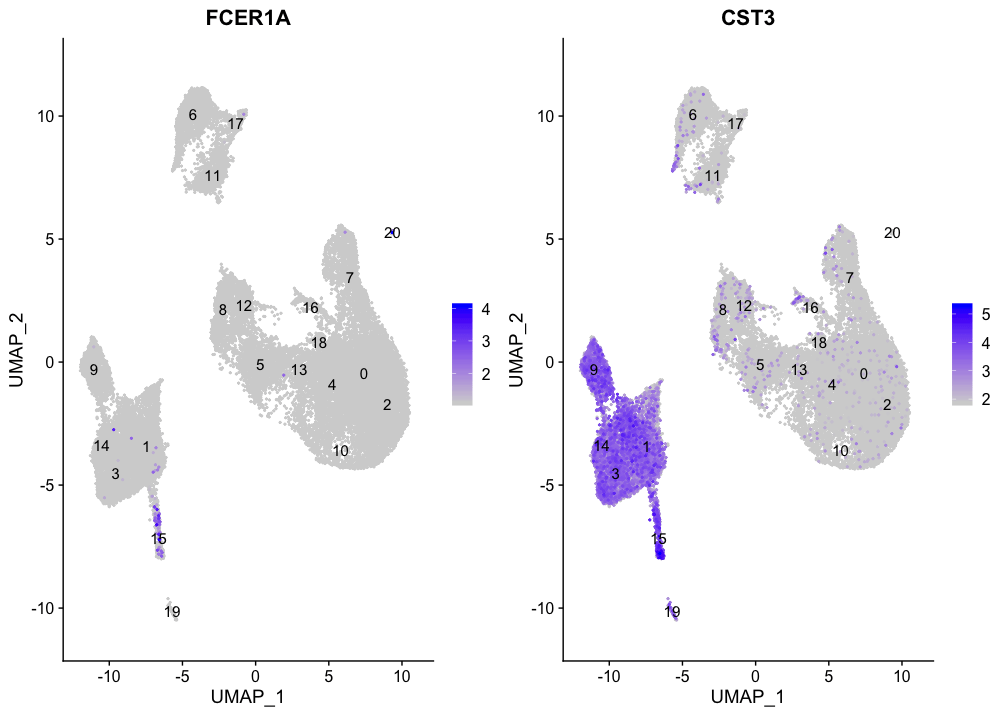
与传统树突状细胞相对应的标记物确定了第15组(两个标记物都持续显示表达)。
浆细胞样树突状细胞标记
FeaturePlot(seurat_integrated,
reduction = "umap",
features = c("IL3RA", "GZMB", "SERPINF1", "ITM2C"),
order = TRUE,
min.cutoff = 'q10',
label = TRUE)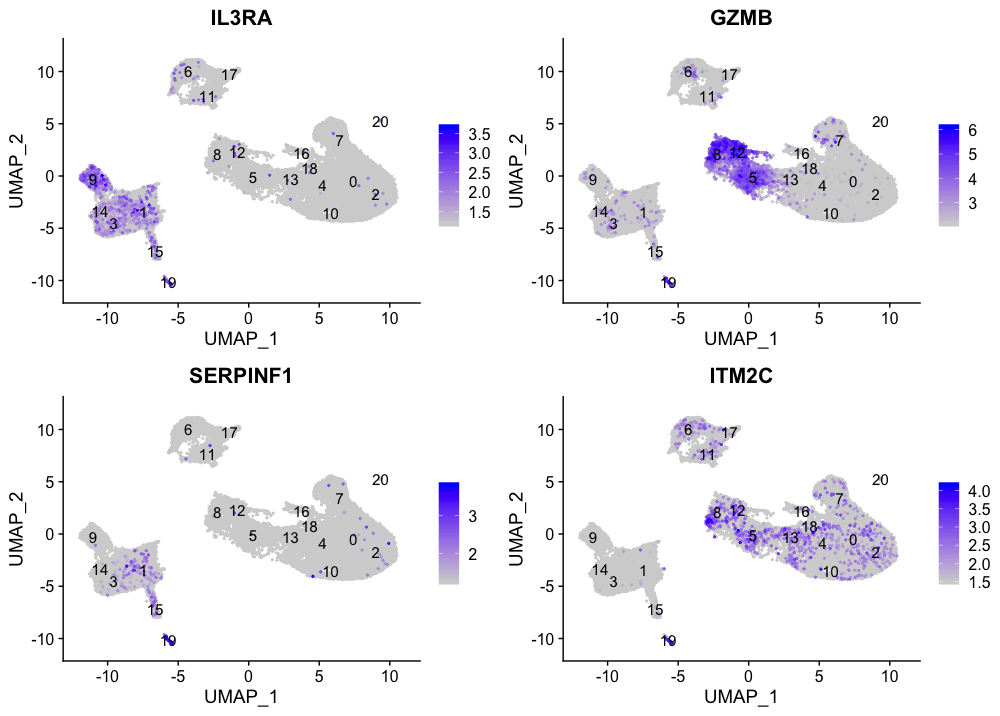
浆细胞树突状细胞代表第19群。虽然这些标记物的表达有很多不同之处,但我们看到第19群持续强烈表达。
如果任何一个聚类似乎包含两种不同的细胞类型,那么提高聚类分辨率以适当地对聚类进行分类是有帮助的。另外,如果我们在提高分辨率的情况下仍然无法分离出这些聚类,那么可能是我们使用的主成分太少,以至于我们没有分离出这些感兴趣的细胞类型。为了告知我们对主成分的选择,我们可以看一下我们的主成分基因表达与UMAP图的重叠情况,并确定我们的细胞群是否被所包含的主成分所分离。



暂无评论内容