

在完成单细胞分析基本流程之后,我们获得了各个细胞聚类和相应的marker基因,有多种方式可以可视化marker基因的表达量,seurat包中自带的DoHeatmap()、VlnPlot()以及DotPlot()函数可以很方便的绘制热图、小提琴图和气泡图,这里主要讲一下热图。
DoHeatmap()绘制出来的热图是这样的:
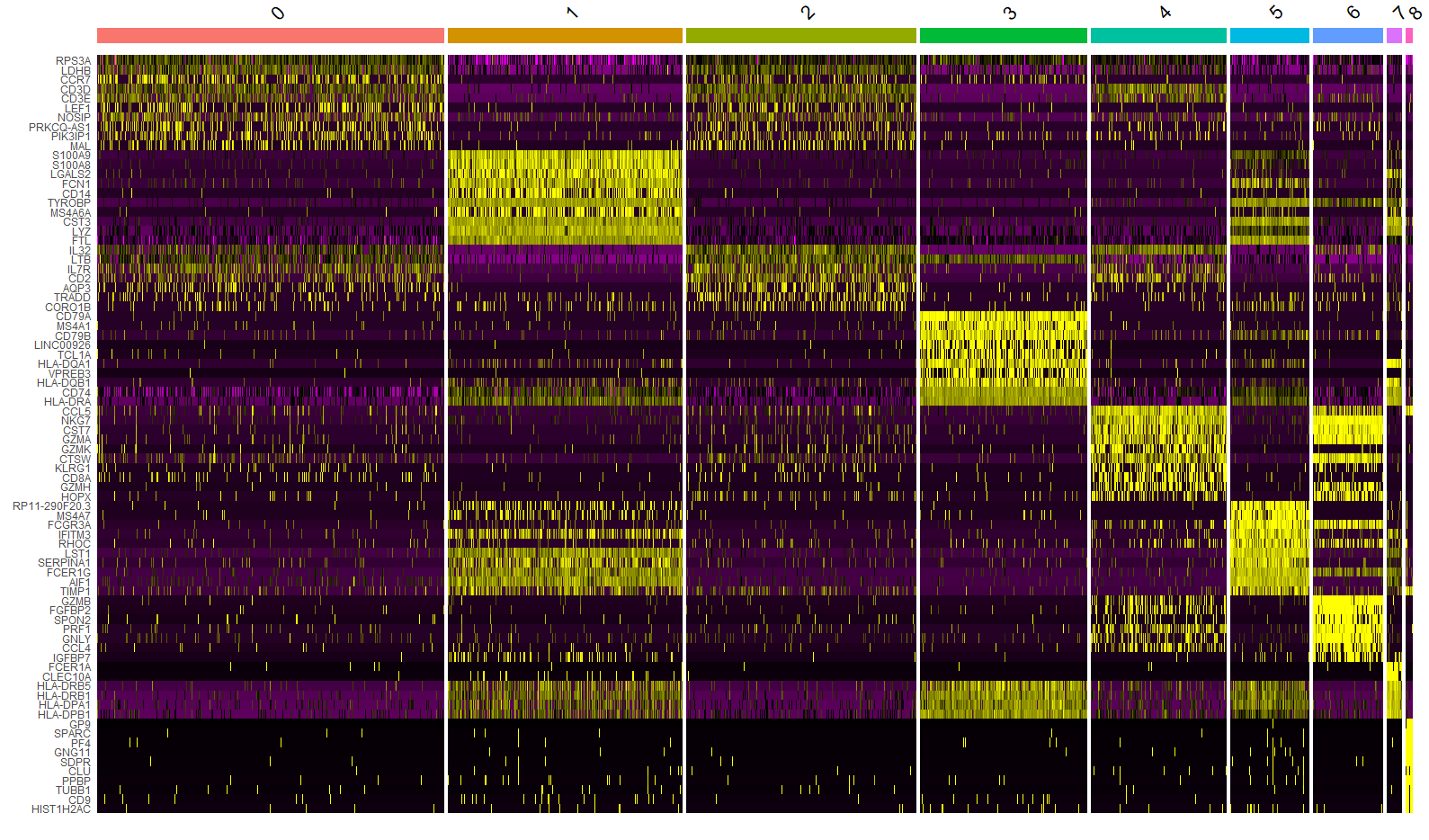
如果前面样本的整合以及聚类分群标志物鉴定都做的比较好的话,整体效果应该还是很不错的,可以明显区分出marker基因在每群细胞中的表达水平。但是个人在使用过程中遇到几个问题:
- 分群数量较多的整张图会很大,完全不适合放在文章里展示,甚至汇报的时候也不方便。
- 将每个细胞的表达量绘制每个格子(cell),会导致细胞数多的分群占的宽,而细胞数少的分群很窄,到后面的分群可能基本看不清,难以放在一起比较。
- 总觉得不太美观。(颜狗🤦♂️)
但是事实上我在人家高分文章中看到的热图一般是这样的:
(虽然该图是作者通过流式分选出各种类型的细胞做的bulk-seq绘制的热图,与单细胞的数据不一样,但是我们还是可以参考一下)
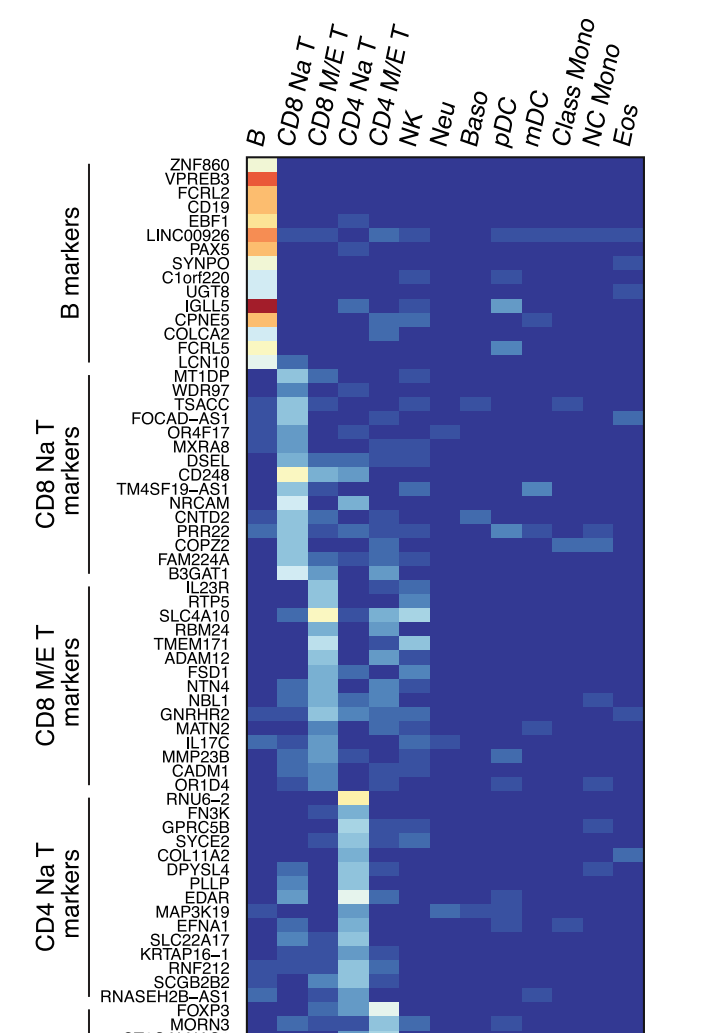
Travaglini, K.J., et al., A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature, 2020. 587(7835): p. 619-625.
或者是这样的:
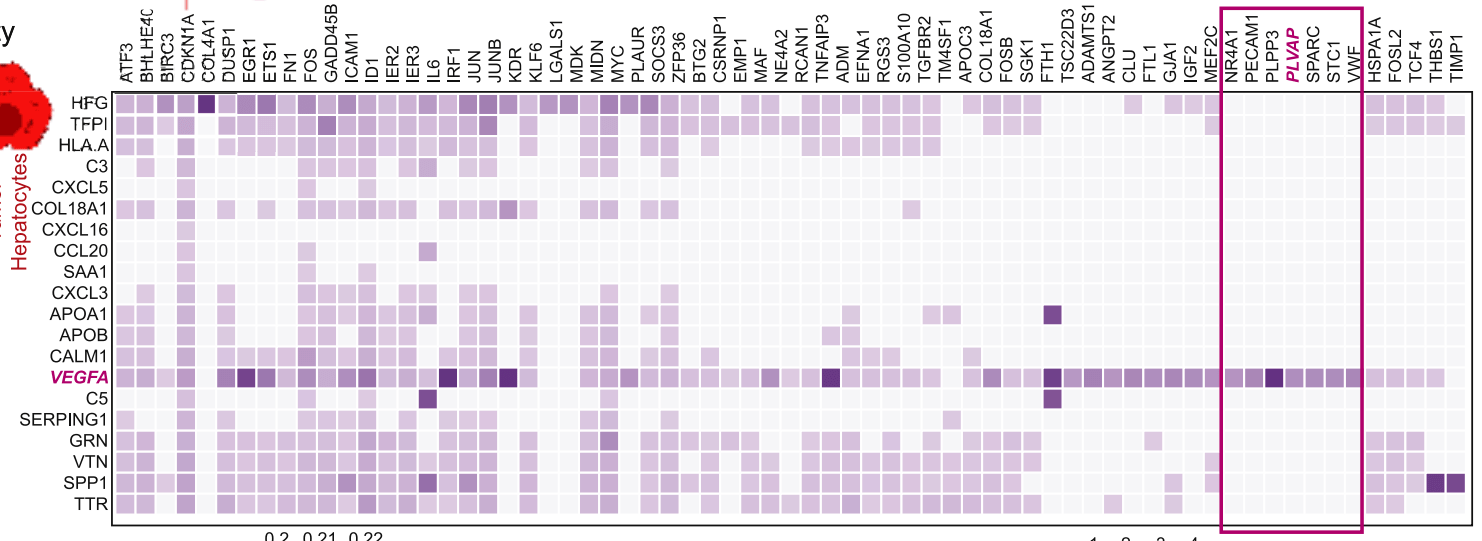
Sharma, A., et al., Onco-fetal Reprogramming of Endothelial Cells Drives Immunosuppressive Macrophages in Hepatocellular Carcinoma. Cell, 2020. 183(2): p. 377-394 e21.
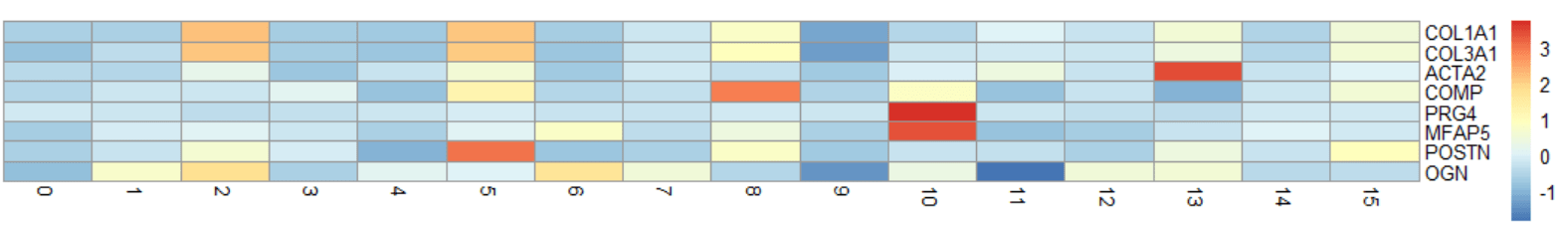
先不论配色如何,他们的共同特点就是热图中每个格子(cell)的色块表示的是某一群细胞(cluster),而非单个的细胞(barcode)。
seurat中恰好有一个函数,可以计算cluster中某基因的平均表达量:AverageExpression()
cluster.averages <- AverageExpression(scRNA,
assays = "RNA",
return.seurat = TRUE,
slot = "data")这里获取scRNA这个对象中RNA槽的data数据,一般已经是经过归一化处理的,其实在seurat中DoHeatmp()函数默认使用的是scale.data数据,目的是为了防止高表达的基因过于凸显,而导致表达量相对较低的基因在聚类间的差异不明显。但是scale之后的数据抹去了基因间的差异,所以有时候要酌情根据自己的需要去选择。
cluster.averages是一个seurat对象,我们也可以在此处再进行scale,这个对象中就同时有了data和scale.data两个slot。不过记住,这里cluster.averages中的rownames已经不是barcode了,cluster的编号或者是指定的细胞类型名称。
cluster.averages <- ScaleData(cluster.averages)
> head(colnames(cluster.averages@assays$RNA@counts))
data[, 1]0 data[, 1]1 data[, 1]2 data[, 1]3 data[, 1]4 data[, 1]5
"0" "1" "2" "3" "4" "5"随意选择想要绘制的基因,当然也可以选择marker基因中获取的top5$gene,就可以绘制热图啦。
genes = c("COL1A1", "COL3A1", "ACTA2", "COMP", "PRG4", "MFAP5", "POSTN", "OGN")
mat <- GetAssayData(cluster.averages, slot = "scale.data")
mat <- as.matrix(mat[genes, ])
library("pheatmap")
pheatmap(mat,
cellheight=10,
cluster_cols=F,
cluster_rows=F,
show_colnames=T)这里绘制热图用的包是pheatmap,自带的配色要好看些,只是要手动提取一下数据。当然也可以使用seurat自带的热图进行绘制。
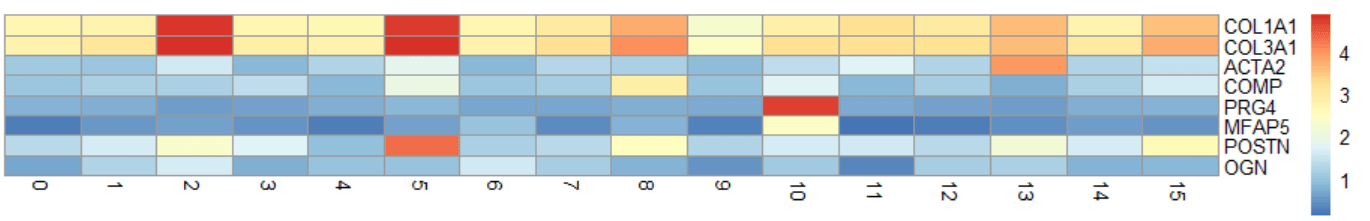
slot = “data”
slot = “scale.data”



暂无评论内容