

在我们可以聚类细胞并识别不同的潜在细胞类型之前,我们还有几个步骤。我们的数据集有两个样本来自两个不同的条件(控制和刺激),所以整合这些样本,以更好地进行比较可能是有帮助的。我们需要将基因表达值归一化,并根据我们数据集中最大的变异来源,对我们的细胞进行跨条件对齐。在本文中,我们将讨论并执行聚类之前的这些初始步骤。
目标:
- 准确地对基因表达值进行归一化和缩放,以解决测序深度的差异和过度分散的计数值。
- 识别出可能表示不同细胞类型的变异最大 (most variant)的基因。
面临的挑战:
- 检查和删除不需要的变异。
建议:
- 在进行聚类之前,对细胞类型有一个明确的预估。知道你期望的细胞类型是否包括低复杂度和线粒体含量较高的细胞类型,以及这些细胞是否正在分化
- 如果需要并适合于实验,排除UMI的数量(默认使用sctransform),线粒体含量和细胞周期的影响,不至于让这些因素影响下游聚类
设置 (Set-up)
为了执行这个分析,我们将主要使用Seurat包中的函数。因此,我们除了需要tidyverse库还需加载Seurat库。创建脚本SCT_integration_analysis.R并加载库。
# 单细胞RNA测序分析 —— 聚类分析
# 加载需要的库
library(Seurat)
library(tidyverse)
library(RCurl)
library(cowplot)为了执行分析,Seurat要求数据必须以seurat对象的形式出现。我们在质控部分中创建了这个对象(filtered_seurat),所以可以直接使用这个对象。
探索不需要变异来源
对生物协变量的校正可用来挑选出特定的特定生物学信号,而对技术协变量的校正对于发现潜在的生物信号可能至关重要。最常见的生物学数据校正是消除细胞周期对转录组的影响。可以通过针对细胞周期得分的简单线性回归来执行此数据校正,这将在下面进行演示。
第一步是探索数据,看看这些协变量对数据是否有影响。原始计数在单元格之间不具有可比性,因此我们无法按原样使用它们进行探索性分析。因此,我们将通过除以每个单元格的总计数并取自然对数来执行粗略的归一化。归一化仅是为了探索我们数据变异的来源。
Seurat最近推出了一种称为sctransform的新标准化方法,该方法可以同时执行方差稳定化并回归出不需要的方差。这是我们在工作流程中实施的规范化方法
# Normalize the counts
seurat_phase <- NormalizeData(filtered_seurat)评估细胞周期对数据的影响
要根据G2 / M和S细胞周期标记物的表达为每个细胞分配分数,我们可以使用Seuart函数CellCycleScoring()。此功能基于输入所需的规范标记来计算细胞周期阶段评分。
一旦数据进行了测序深度的归一化,我们就可以根据每个细胞的G2/M和S期标志物的表达情况,给每个细胞分配一个分数。
我们提供了一份人类细胞周期标志物列表,供大家下载。在这个链接上右键点击 "将链接保存为…",直接进入你的data目录。
如果下载速度过慢,可尝试: https://vamond.lanzous.com/ijxLJoe98xi
密码:6qfr
# 导入细胞周期标记物
load("data/cycle.rda")
# 给细胞的细胞周期评分
seurat_phase <- CellCycleScoring(seurat_phase,
g2m.features = g2m_genes,
s.features = s_genes)
# 观察细胞周期评分和分配给每个细胞的周期
View(seurat_phase@meta.data)在对细胞周期进行评分后,我们想用PCA来确定细胞周期是否是我们数据集中变异的主要来源。要执行PCA,我们要先识别高度变异基因,然后对数据进行缩放。由于高表达的基因表现出最高的变异量,我们不希望我们的 "高变异基因 "只反映高表达,所以我们需要对数据进行缩放,以缩放变异与表达水平。Seurat的ScaleData()函数将通过以下方式对数据进行缩放。
- 调整每个基因的表达量,使每个基因在细胞间的平均表达量为0。
- 缩放每个基因的表达量,以使得跨细胞的方差为1。
# 找出变异最大的2000个基因
seurat_phase <- FindVariableFeatures(seurat_phase,
selection.method = "vst",
nfeatures = 2000,
verbose = FALSE)
# 缩放计数
seurat_phase <- ScaleData(seurat_phase)现在,我们可以进行PCA分析,并对最重要的的PC进行绘图。
# 运行 PCA
seurat_phase <- RunPCA(seurat_phase)
# 使用细胞周期阶段对PCA进行着色
DimPlot(seurat_phase,
reduction = "pca",
group.by= "Phase",
split.by = "Phase")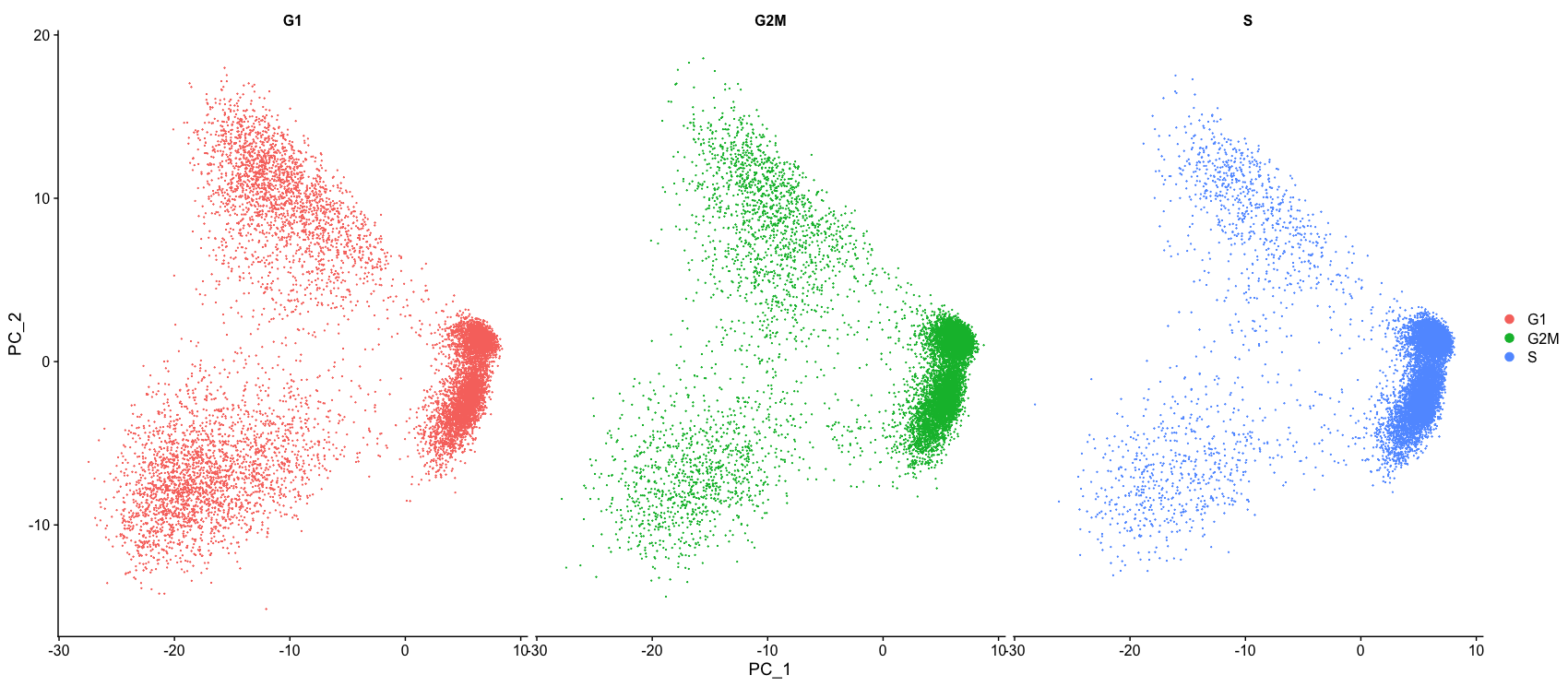
什么情况下需要对细胞周期引起的变异进行退回?
下面是从Seurat官方教程中获得的两个PCA图(“细胞周期评分和回归”)。
第一张图类似于我们上面绘制的图,它是回归之前的PCA,用于评估细胞周期是否在驱动PC1和PC2中发挥重要作用。
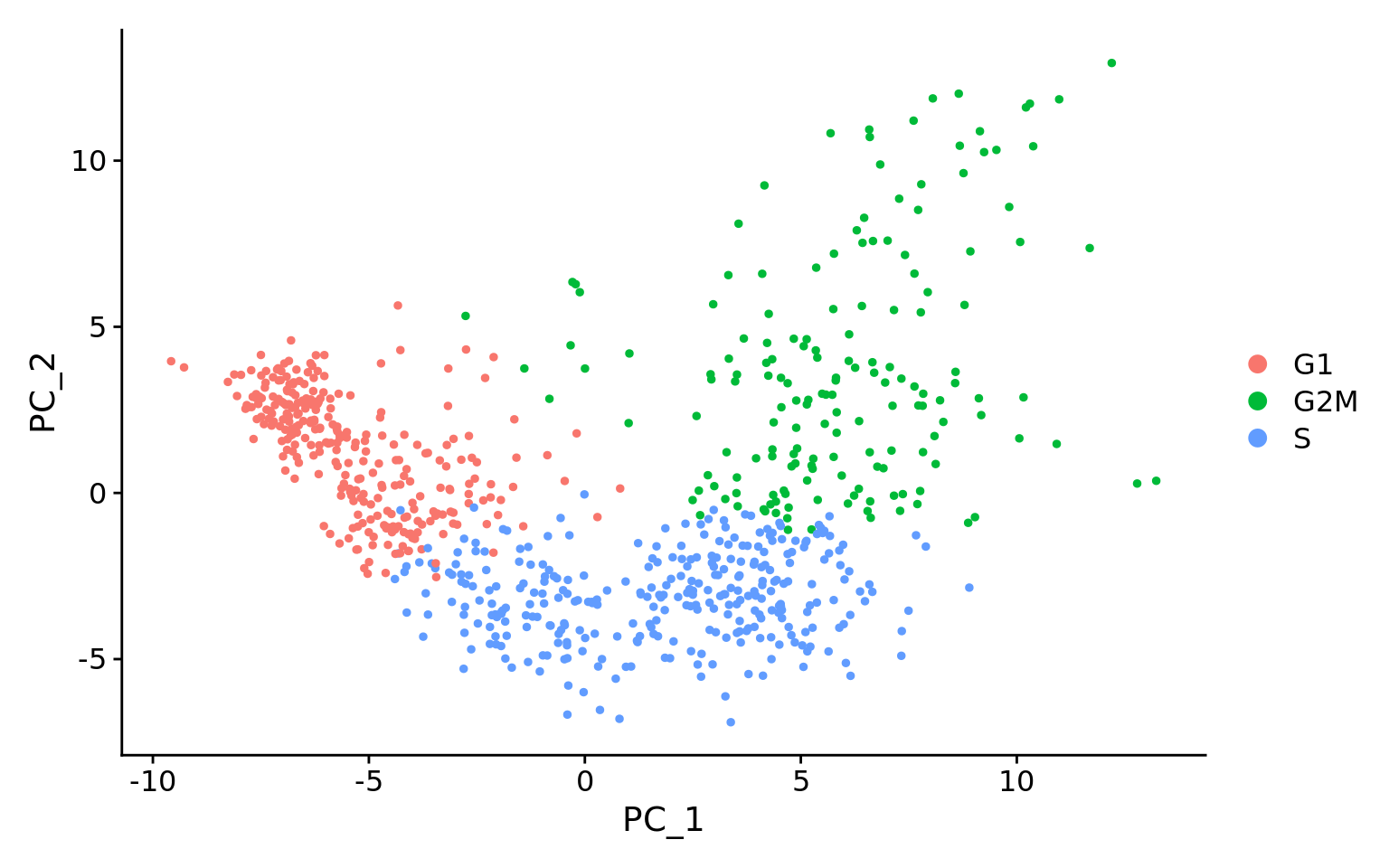
显然,在这种情况下,细胞是按细胞类型分隔的,因此该插图建议消除这些影响。
这第二个PCA图是回归后的,它显示了回归在消除我们观察到的效果方面的效果。
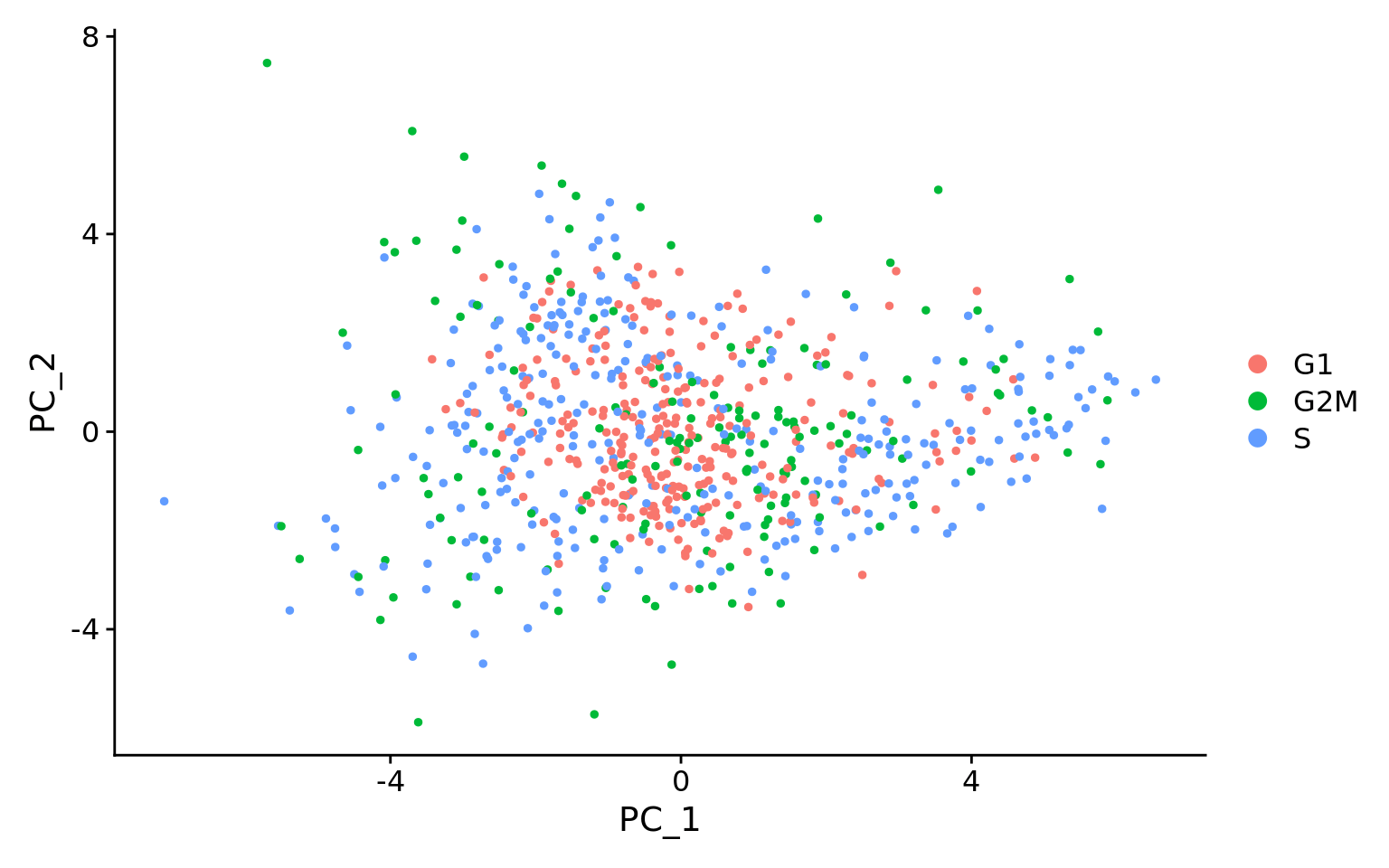
评估线粒体表达的影响
线粒体表达是另一个可以极大地影响聚类的因素。通常,将由于线粒体表达而引起的变异退回非常有用。但是,如果线粒体基因表达的差异代表可能有助于区分细胞簇的生物学现象,那么我们建议不要将其退步。我们可以执行类似于查看细胞周期的快速检查,但是我们首先可以将线粒体比率变量转换为基于四分位数的分类变量。
# Check quartile values
summary(seurat_phase@meta.data$mitoRatio)
# Turn mitoRatio into categorical factor vector based on quartile values
seurat_phase@meta.data$mitoFr <- cut(seurat_phase@meta.data$mitoRatio,
breaks=c(-Inf, 0.0144, 0.0199, 0.0267, Inf),
labels=c("Low","Medium","Medium high", "High"))
# Plot the PCA colored by mitoFr
DimPlot(seurat_phase,
reduction = "pca",
group.by= "mitoFr",
split.by = "mitoFr")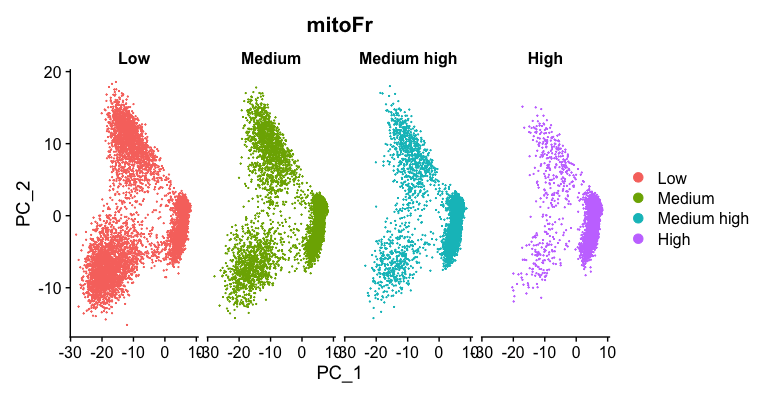
这个图我自己做出来的跟教程中给的示例是不一样的
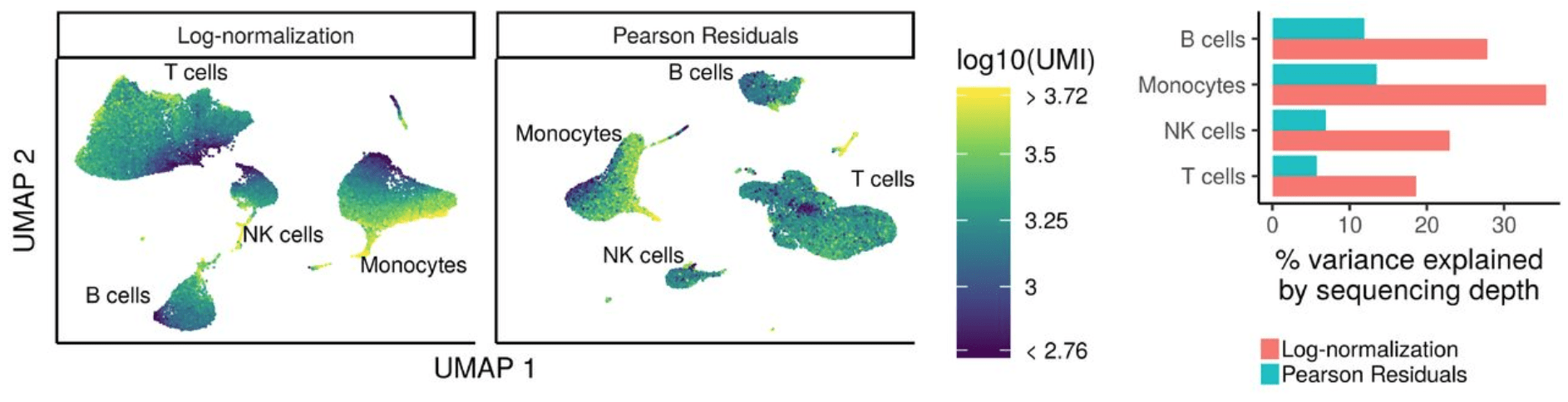
基于此图,我们可以看到该图包含具有“高”线粒体表达细胞的图,具有不同的散布模式。我们观察到,图左侧的细胞瓣是大多数具有高线粒体表达的细胞所在的位置。对于线粒体表达的所有其他水平,我们看到整个PCA图中的细胞分布更加均匀。因为我们看到了明显的差异,所以当我们识别出变异最大的基因时,我们将回归“ mitoRatio”。
归一化并去除不需要的变异
现在,我们可以将sctransform方法用作进行归一化,估计原始过滤数据的方差并确定变化最大的基因的更准确方法。 sctransform方法使用正则化的负二项式模型对UMI计数进行建模,以消除由于测序深度(每个细胞的总nUMI)所引起的变异,同时基于具有相似丰度的基因之间的合并信息来调整方差(类似于一些大体积RNA-seq方法)。
Hafemeister C and Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression, bioRxiv 2019 (https://doi.org/10.1101/576827)
模型的输出(残差)是测试的每个转录本的标准化表达水平。
Sctransform通过回归测序深度(nUMI)自动计算细胞测序深度。但是,如果在探索步骤中发现了其他有趣的数据变化来源,我们也可以将其包括在内。由于细胞周期阶段,我们观察到几乎没有影响,因此我们选择不将其从我们的数据中去除。我们观察到了线粒体表达的一些影响,因此我们选择从数据中回归出来。
为了运行SCTransform,我们以下面的代码为例。不要运行此代码,因为我们希望在下面的下一部分中为每个示例分别运行此代码。
## DO NOT RUN CODE ##
# SCTranform
seurat_phase <- SCTransform(seurat_phase, vars.to.regress = c("mitoRatio"))遍历数据集中的样本
由于我们的数据集中有两个样本(来自两个条件),因此我们希望将它们保留为单独的对象并进行转换,之后才可以进行整合。我们将首先将seurat_phase对象中的单元格拆分为“Control”和“Stimulated”:
# Split seurat object by condition to perform cell cycle scoring and SCT on all samples
split_seurat <- SplitObject(seurat_phase, split.by = "sample")
split_seurat <- split_seurat[c("ctrl", "stim")]现在,我们将使用“ for循环”在每个样本上运行SCTransform(),并通过在SCTransform()函数的vars.to.regress参数中指定来回归线粒体表达式。
在我们运行这个for循环之前,我们知道在内存方面,输出可以产生大的R对象变量。如果我们有一个大的数据集,那么我们可能需要使用以下代码调整R内部允许的对象大小的限制(默认是500 * 1024 ^ 2 = 500 Mb)。
options(future.globals.maxSize = 4000 * 1024^2)现在,我们运行以下循环以对所有样本执行sctransform。这可能需要一些时间:
for (i in 1:length(split_seurat)) {
split_seurat[[i]] <- SCTransform(split_seurat[[i]], vars.to.regress = c("mitoRatio"))
}默认情况下,在归一化,调整方差并消退无用的变异源之后,SCTransform将按残差对基因进行排序,并输出3000个最大变异的基因。如果数据集中细胞数量较多,则使用variable.features.n参数将此参数调整得更高可能是有益的。
注意,输出的最后一行指定“Set default assay to SCT”。我们可以查看存储在seurat对象中的不同assay。
# Check which assays are stored in objects
split_seurat$ctrl@assays现在我们可以看到,除了原始RNA计数之外,我们的检测槽中现在还有一个SCT组件。最具可变性的特征将是SCT分析中存储的唯一基因。随着我们对scRNA-seq分析的进行,我们将选择最合适的分析方法用于分析中的不同步骤。
思考题
- 是否可以对split_seurat对象中的“刺激”样本使用相同的检测方法?您用来检查的代码是什么?
- 对于“ ctrl”和“ stim”相对于“前10个特征:”和“前10个可变特征:”中列出的基因或特征有何发现?
保存对象
在结束之前,让我们将该对象保存到data /文件夹中。可能需要一段时间才能返回到此阶段,尤其是在处理大型数据集时,最佳做法是将对象另存为本地易于加载的文件。
# Save the split seurat object
saveRDS(split_seurat, "data/split_seurat.rds")要将.rds文件加载回您的环境,请使用以下代码:
# Load the split seurat object into the environment
split_seurat <- readRDS("data/split_seurat.rds")点击此链接即可访问原文



暂无评论内容