

参考链接: https://satijalab.org/seurat/articles/pbmc3k_tutorial.html
建立 Seurat 对象
示例数据为10X Genomics的外周血单个核细胞(PBMC)数据集,含有2700个单细胞,使用Illumina NextSeq 500平台测得。点击原始数据下载。
首先使用Read10X()函数可以直接读取cellranger的输出文件夹得到一个UMI count 矩阵(a unique molecular identified (UMI) count matrix)。矩阵内的值表示每个feature(基因,行)在每个细胞(列)中的表达。
然后使用这个count矩阵创建Seurat对象,用来储存原始数据(count 矩阵)和分析结果(比如PCA分析和聚类的结果等)。
比如,此数据集中,count矩阵储存在pbmc[["RNA"]]@counts中。
R语言读取文件路径只认识反斜线 “/”,回车键左边那个 (从windows文件属性复制过来的需要手动修改)
library(dplyr)
library(Seurat)
library(patchwork)
# Load the PBMC dataset
pbmc.data <- Read10X(data.dir = "../data/pbmc3k/filtered_gene_bc_matrices/hg19/")
# Initialize the Seurat object with the raw (non-normalized data).初始化 Seurat 对象,保留至少在三个细胞中表达的基因,保留至少包含 200 个基因的细胞(初步的筛选)
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
pbmc可以看到pbmc这个seurat对象的摘要信息:2700个样本(细胞),通过RNA测到的13714个特征(基因)
> pbmc
An object of class Seurat
13714 features across 2700 samples within 1 assay
Active assay: RNA (13714 features, 0 variable features)我们看一下前三十个细胞中的几个基因表达的情况。c()表示一个“向量“,可以理解成是列表,组合。sparse Matrix是稀疏矩阵(对应R语言中的对象类型dgCMatrix),由于单细胞测序的数据中含有很多0,使用稀疏矩阵可以降低文件的大小,提高分析效率,程序输出的.就是0,即未测得。 ‘AAACATACAACCAC-1’, ‘AAACATTGAGCTAC-1’, ‘AAACATTGATCAGC-1’ ...是每个细胞的标记barcode,测序中仪器给每个细胞生成的特异性识别序列。
> pbmc.data[c("CD3D", "TCL1A", "MS4A1"), 1:30]
3 x 30 sparse Matrix of class "dgCMatrix"
[[ suppressing 30 column names ‘AAACATACAACCAC-1’, ‘AAACATTGAGCTAC-1’, ‘AAACATTGATCAGC-1’ ... ]]
CD3D 4 . 10 . . 1 2 3 1 . . 2 7 1 . . 1 3 . 2 3 . . . . . 3 4 1 5
TCL1A . . . . . . . . 1 . . . . . . . . . . . . 1 . . . . . . . .
MS4A1 . 6 . . . . . . 1 1 1 . . . . . . . . . 36 1 2 . . 2 . . . .# 可见稀疏矩阵空间占用更低
> dense.size <- object.size(as.matrix(pbmc.data))
> dense.size
709591472 bytes
> sparse.size <- object.size(pbmc.data)
> sparse.size
29905192 bytes
> dense.size/sparse.size
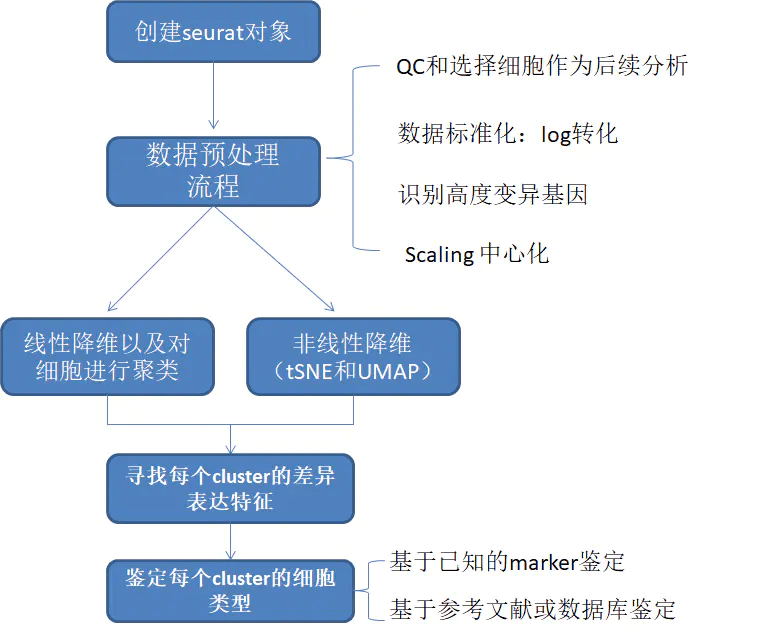
23.7 bytes标准预处理流程
质控(QC)选择细胞进行后续分析
常用的QC指标:
- 每个细胞检测到的基因数
- 低质量的细胞或空的droplets一般只会被检测到较少的基因
- doublets或multiplets会有异常高的基因数
- 每个细胞检测到的分子总数 total molecules(与该细胞中测到的基因数 unique genes 应该是正相关的)
- 比对到线粒体基因组的reads比例
- 状态很差以及死细胞中通常会包含大量的线粒体污染
- 可以基于线粒体QC矩阵使用
PercentageFeatureSet()函数计算每个细胞的线粒体基因的百分比 - 以
MT-开头的基因名就是来自线粒体的基因(有些数据是以小写mt-开头的)
# 用 [[ 可以在对象的 metadata 中添加列
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")seurat对象中QC参数的储存位置
- 在使用
CreatSeuratObject()函数的时候会自动计算 total molecules 和 unique genes
并且保存到seurat对象的meta.data这个slot里面:
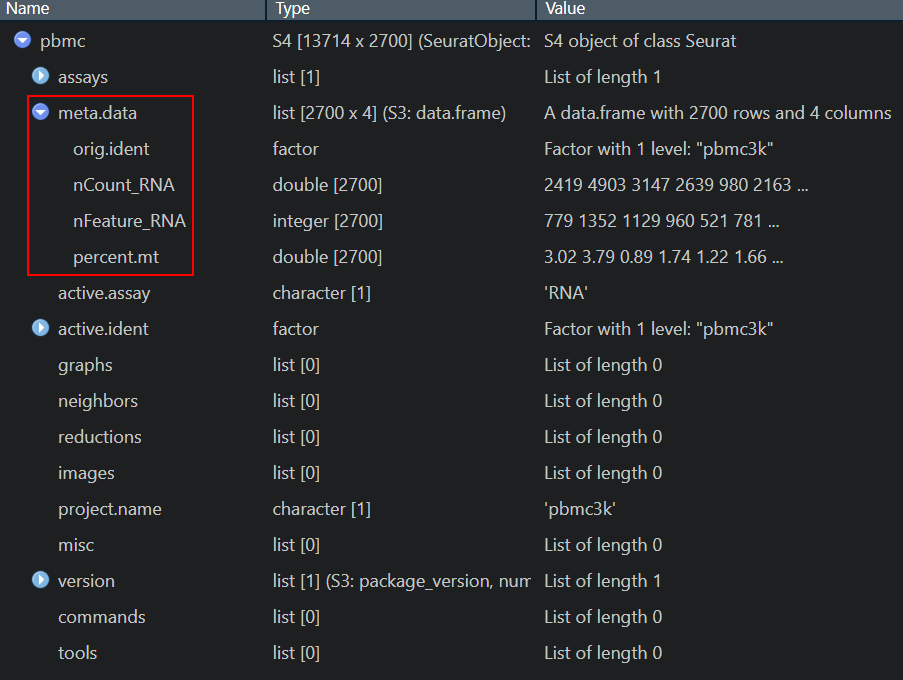
# Show QC metrics for the first 5 cells
> head(pbmc@meta.data, 5)
orig.ident nCount_RNA nFeature_RNA percent.mt
AAACATACAACCAC-1 pbmc3k 2419 779 3.0177759
AAACATTGAGCTAC-1 pbmc3k 4903 1352 3.7935958
AAACATTGATCAGC-1 pbmc3k 3147 1129 0.8897363
AAACCGTGCTTCCG-1 pbmc3k 2639 960 1.7430845
AAACCGTGTATGCG-1 pbmc3k 980 521 1.2244898head()/tail()函数取对象的最前/最后x个元素
@获取对象的slot,$获取变量
- 可以使用seurat将QC参数可视化,进一步用来筛选细胞:
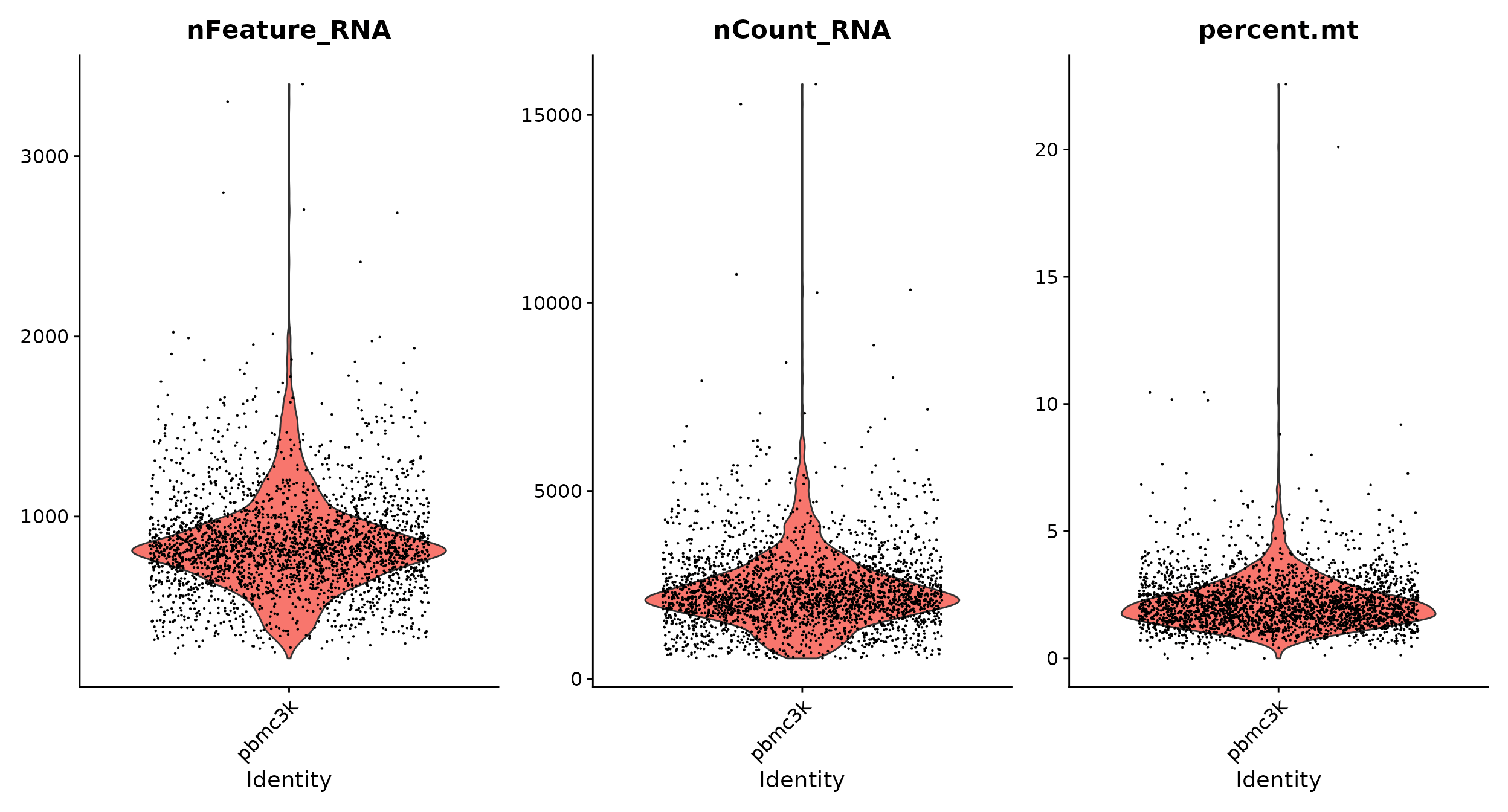
# 使用小提琴图可视化QC参数
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)VlnPlot()函数第一个参数是seurat对象名称,features指定要绘图的参数,ncol表示分几列显示。
nFeature_RNA就是每个细胞检测到的基因数目,就是以前版本的nGene;nCount_RNA就是这些基因数目一共测到的count数目,也就是以前版本的UMI数目;percent.mt(前面使用PercentageFeatureSet()函数计算线粒体QC指标,该函数计算源自一组特征的计数百分比)
# 可以用 FeatureScatter 绘制散点图可视化特征之间的关系
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
# CombinePlots(plots = list(plot1, plot2)) 该函数未来将被移除
# 使用patchwork包中的功能合并图片:
plot1 + plot2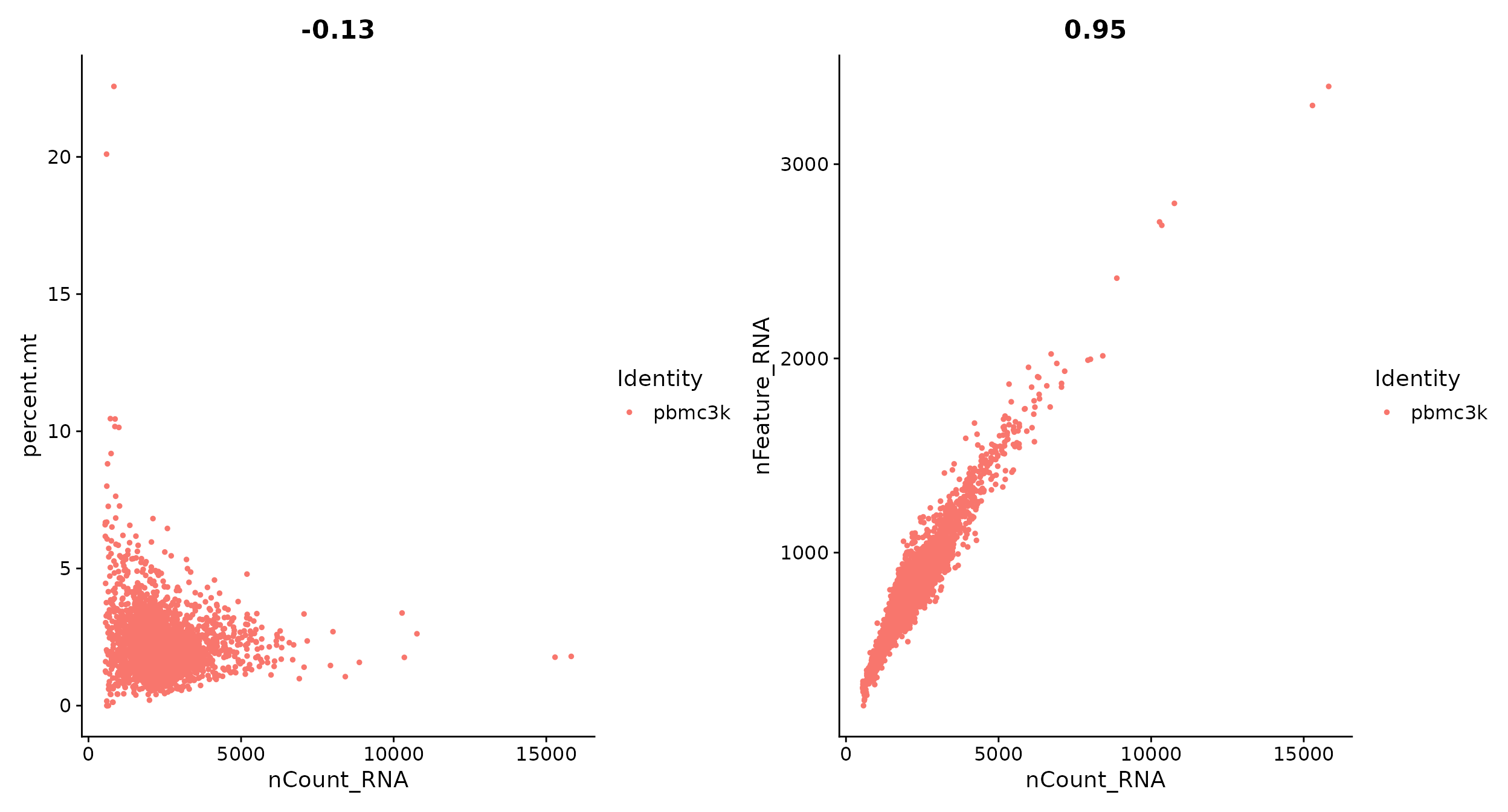
图上方的数字是相关系数
- 去除测到基因数(unique feature counts)大于2500的以及小于200的细胞
- 去除线粒体基因counts比例大于5%的细胞
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)运行以后可以看到pbmc中的细胞数从2700变成了2638
数据归一化
数据归一化消除数据量纲之间的不同;消除数量级之间差别很大;避免数值问题:太大的数会引发数值问题;平衡各特征的贡献;一些模型求解的需要:加快了梯度下降求最优解的速度等需要。参考 “归一化”与“标准化”
通过QC筛掉我们不要的细胞后,接下来对数据进行归一化处理。Seurat中默认采用全局归一化方法LogNormalize:通过总表达值对每个cell的表达值进行归一化,将其乘以比例因子(默认为10,000),并对结果进行对数转换。 归一化值存储在pbmc [[“RNA”]] @ data中。
#使用log转化,度量单位是10000
pbmc <- NormalizeData(object = pbmc, normalization.method = "LogNormalize", scale.factor = 1e4)# 归一化以后使用view函数查看一下结果
> view(pbmc[["RNA"]]@data)
Showing first 1000 rows.
识别高度变异基因
寻找出细胞间表达高度变异的基因构建数据子集,在下游分析中会用到(如PCA)。这些基因可能参与重要的生物学信号转导等。在Seurat中使用FindVariableFeatures()函数可以很方便的找到这些高度变异基因。每个数据集默认返回2000个基因。
# 找到2000个高度变异基因
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
# 获取前10个高度变异基因
top10 <- head(VariableFeatures(pbmc), 10)
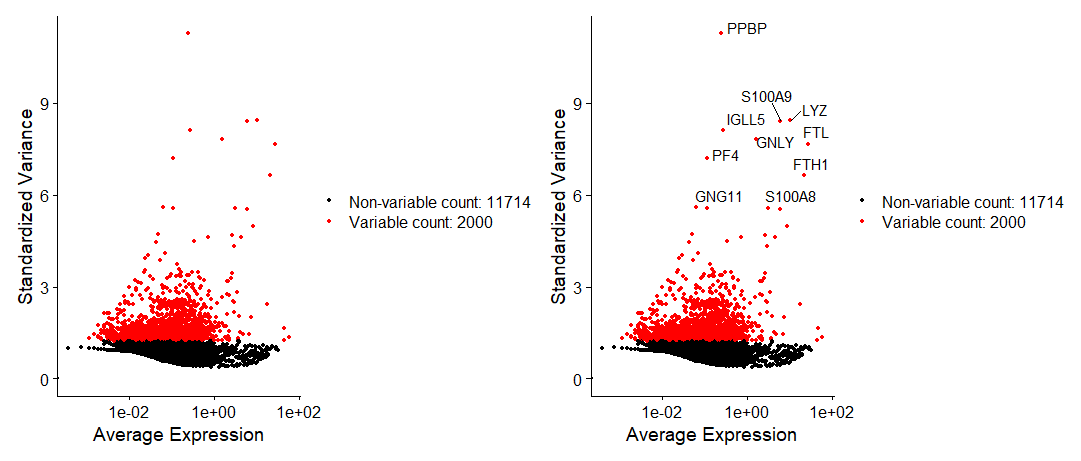
# 分别做一个不带标签和带标签的图看一下
plot1 <- VariableFeaturePlot(pbmc)
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
plot1 + plot2-
运行时遇到一个小issue,做个记录:
分别绘图plot1和plot2都没问题,并且使用
show()也可以显示出来,但是合并图片时出现error> plot1 + plot2 Error in grid.Call(C_convert, x, as.integer(whatfrom), as.integer(whatto), : Viewport has zero dimension(s)后来发现是右下角图片预览窗口太小了无法绘制,将该窗口调整一下重新运行即可。
# 可以查看一下变异基因结果
> view(pbmc$"RNA"@var.features)
Showing first 1000 rows.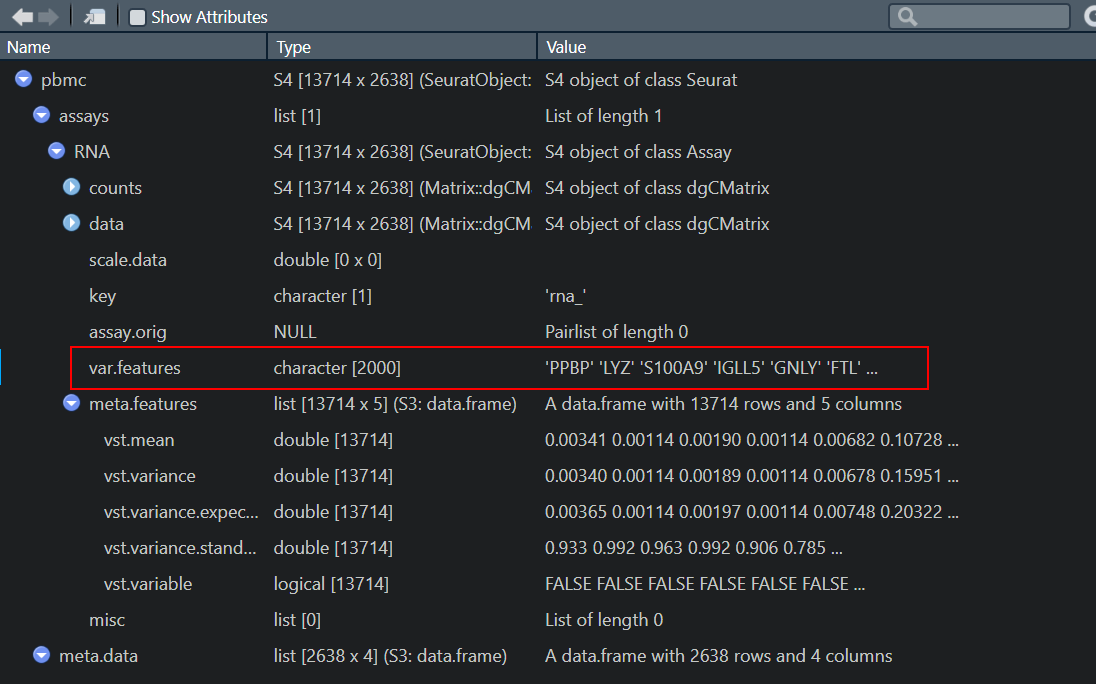
数据标准化、中心化 Scaling the data
应用线性变换(’缩放’),这是在PCA等降维技术之前的标准预处理步骤(目的:将样本中的每个观测值都减掉样本均值,这样做的好处是能够使得求解协方差矩阵变得更容易。)参考 “归一化”与“标准化”
对数据进行标准化后:
- 使每个基因在所有细胞的表达量均值为 0
- 使每个基因在所有细胞中的表达量方差为 1,这一步在下游分析中给予相同权重,高表达基因不占优势。
- 结果储存在
pbmc[["RNA"]]@scale.data
# 数据缩放
all.genes <- rownames(pbmc)
pbmc <- ScaleData(pbmc, features = all.genes)> pbmc <- ScaleData(pbmc, features = all.genes)
Centering and scaling data matrix
|===================================================================================| 100%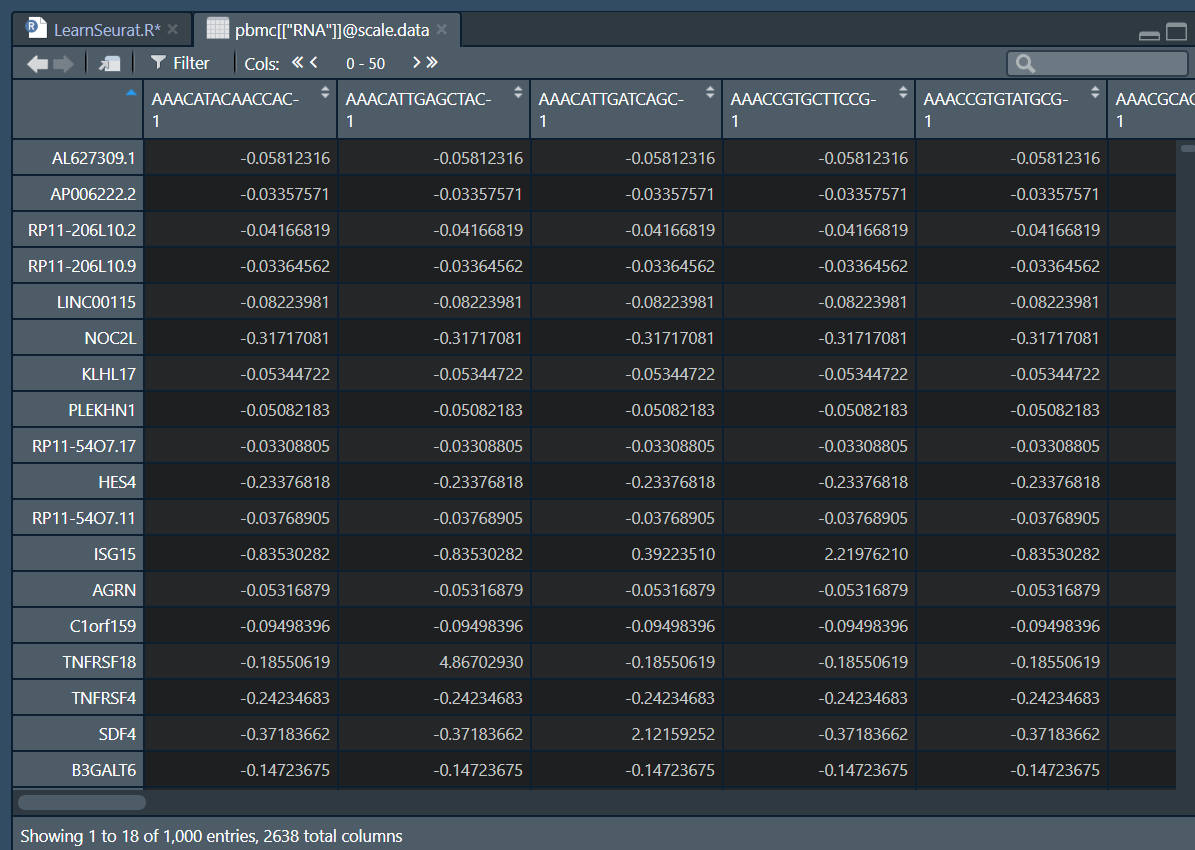
-
数据量大的时候这一步会比较慢,如何加快处理速度?
缩放是seurat进行后续分析的必要步骤,但是只有上文中得到的高度变异的基因会被用于PCA和聚类分析,所以可以不用处理allgenes。
pbmc <- ScaleData(pbmc)由于下文中涉及Seurat中的热图
DoHeatmap()绘制,最好还是将allgenes都缩放一下,以免高表达基因在热图中过于明显。 -
How can I remove unwanted sources of variation, as in Seurat v2?
In Seurat v2 we also use the
ScaleData()function to remove unwanted sources of variation from a single-cell dataset. For example, we could ‘regress out’ heterogeneity associated with (for example) cell cycle stage, or mitochondrial contamination. These features are still supported inScaleData()in Seurat v3, i.e.:pbmc <- ScaleData(pbmc, vars.to.regress = "[percent.mt](http://percent.mt/)")However, particularly for advanced users who would like to use this functionality, we strongly recommend the use of our new normalization workflow,
SCTransform(). The method is described in our paper, with a separate vignette using Seurat v3 here. As withScaleData(), the functionSCTransform()also includes a vars.to.regress parameter.
线性降维 PCA(主成分分析)
通常情况下只有前面找到的高变基因会纳入PCA分析,但是我们也可以使用features参数来选择特定的子集;npcs参数表示计算并储存多少个PCs,默认为50.
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))Seurat提供了多种方式可视化探索PCA数据,包括VizDimReduction(), DimPlot()和DimHeatmap()
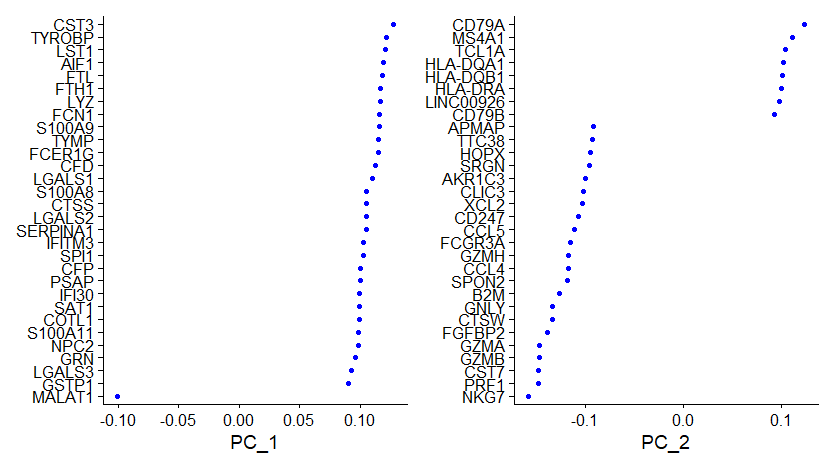
# 检查一下PCA结果
> print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)
PC_ 1
Positive: CST3, TYROBP, LST1, AIF1, FTL
Negative: MALAT1, LTB, IL32, IL7R, CD2
PC_ 2
Positive: CD79A, MS4A1, TCL1A, HLA-DQA1, HLA-DQB1
Negative: NKG7, PRF1, CST7, GZMB, GZMA
PC_ 3
Positive: HLA-DQA1, CD79A, CD79B, HLA-DQB1, HLA-DPB1
Negative: PPBP, PF4, SDPR, SPARC, GNG11
PC_ 4
Positive: HLA-DQA1, CD79B, CD79A, MS4A1, HLA-DQB1
Negative: VIM, IL7R, S100A6, IL32, S100A8
PC_ 5
Positive: GZMB, NKG7, S100A8, FGFBP2, GNLY
Negative: LTB, IL7R, CKB, VIM, MS4A7VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")VizDimLoadings(): Visualize top genes associated with reduction components。dims指定显示的维度,reduction指定降维的方式,结果如下图:
DimPlot(pbmc, reduction = "pca")DimPlot(): Graphs the output of a dimensional reduction technique on a 2D scatter plot where each point is a cell and it's positioned based on the cell embeddings determined by the reduction technique. By default, cells are colored by their identity class (can be changed with the group.by parameter).
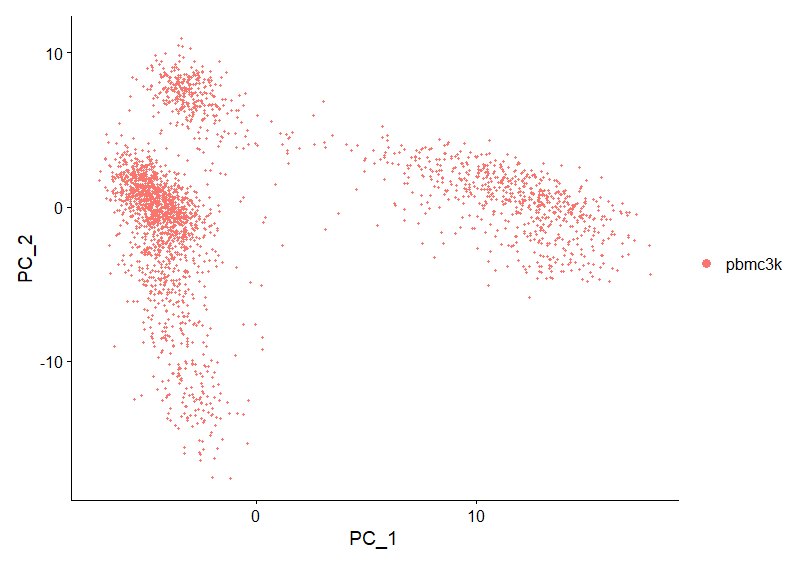
DimHeatmap(pbmc, dims = 1, cells = 500, balanced = TRUE)
# DimHeatmap(pbmc, dims = 1:15, cells = 500, balanced = TRUE)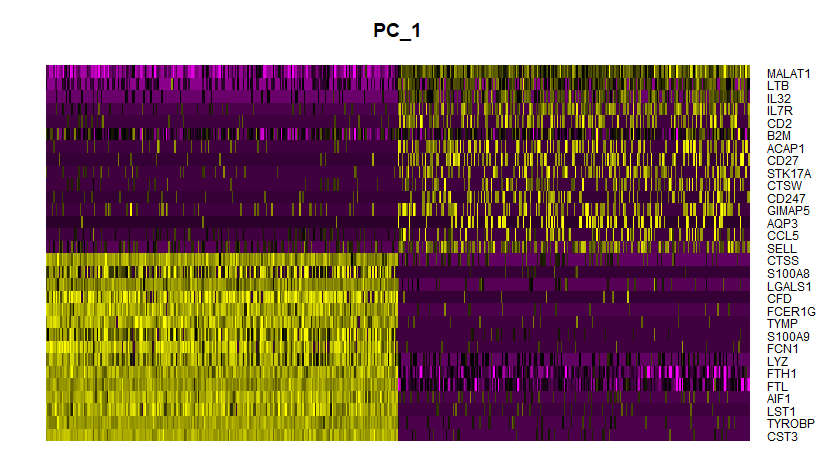
确定数据集的维度
对于单细胞测序中单个基因的数据,会存在大量的技术噪点,Seurat基于PCA结果对细胞进行聚类,每个主成分(PC)代表一个系列的关联基因,最主要的主成分则可以认为是对整个数据集的稳定的压缩。但是我们应该纳入多少主成分进行分析呢?10?20?100?
方法一:JackStraw
随机置换一部分数据(默认为1%),然后重新 PCA,重复此过程。将包含较多低 P 值特征的主成分为「重要的」主成分。
JackStraw()函数可以计算出每个主成分中各基因的P值,用于判断哪些主成分更具有统计学意义,ScoreJackStraw()用于量化主成分的显著性强度,富含低P值基因较多的主成分更有统计学意义。
# NOTE: 对于比较大的数据集 JackStraw 比较耗时More
# approximate techniques such as those implemented in ElbowPlot() can be used to reduce
# computation time
pbmc <- JackStraw(pbmc, num.replicate = 100)
pbmc <- ScoreJackStraw(pbmc, dims = 1:20)
JackStrawPlot()函数可视化比较每个主成分的 p 值分布和均匀分布(虚线)。在这个例子中,在前 10 到 12 个主成分之后,主成分的重要性开始下降。
JackStrawPlot(pbmc, dims = 1:15)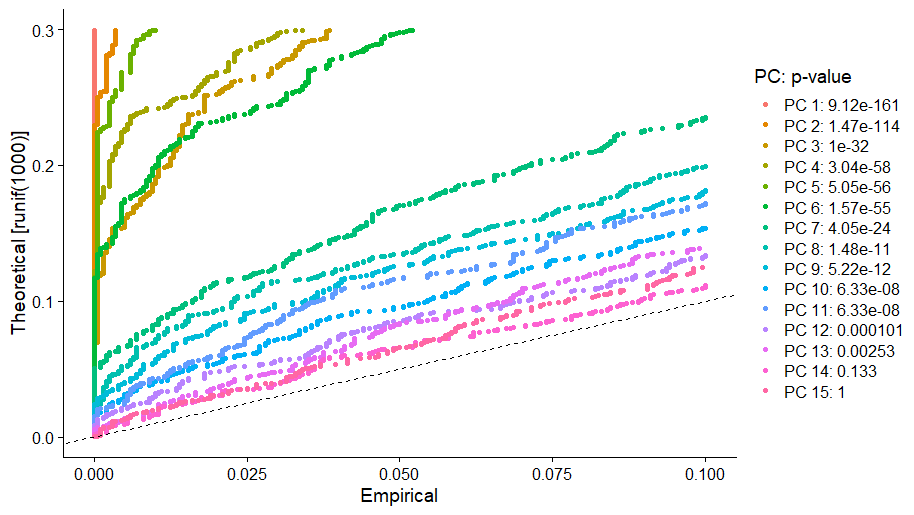
方法二:碎石图 Elbow plot
ElbowPlot():a ranking of principle components based on the percentage of variance explained by each one.
本例中我们可以看到在9~10主成分出现转角,表明真实的信号主要来自前10个主成分
ElbowPlot(pbmc)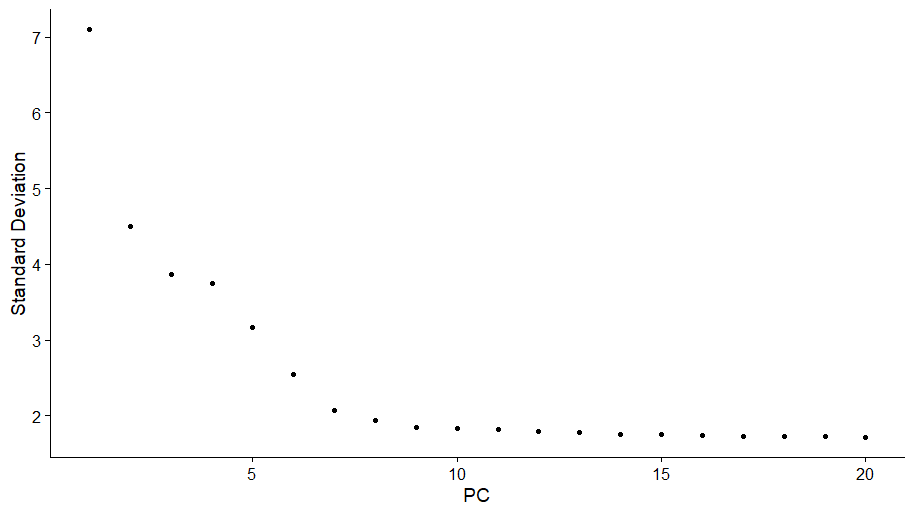
关于数据集维度的选择
本例选择10 ,但是确认数据维度通常比较麻烦,seurat官方教程推荐考虑以下三个方法:
- 树突状细胞和NK细胞研究会发现本数据集中PCs12和PCs13内的基因与一些含量较低的免疫细胞相关(如MZB1是浆细胞样树突状细胞的标记物)。但是这些细胞群含量极低,在没有先验知识的前提下,由于此例的样本量较低,很难与背景噪音区分开来。
- 推荐尝试使用不同PCs数进行下游分析(10,15甚至50!)多数情况下其结果不会有显著的差异。
- 推荐选择相对较大的数,比如在此例中,如果只选择PCs数为5,最终的结果会有显著差异,甚至相反。
细胞聚类 Cluster the cells
首先基于 PCA 空间中的欧式距离构造 K-nearest neighbor(KNN) 图,然后基于其局部邻域中的共享重叠来细化任意两个细胞之间的距离权重。(输入之前定义的数据集维度)
接着优化模型,resolution 参数决定下游聚类分析得到的分群数,对于 3K 左右的细胞,建议设为 0.4-1.2 ;如果数据量增大,该参数也应增大。
pbmc <- FindNeighbors(pbmc, dims = 1:10)
pbmc <- FindClusters(pbmc, resolution = 0.5)# Look at cluster IDs of the first 5 cells
> head(Idents(pbmc), 5)
AAACATACAACCAC-1 AAACATTGAGCTAC-1 AAACATTGATCAGC-1 AAACCGTGCTTCCG-1
2 3 2 1
AAACCGTGTATGCG-1
6
Levels: 0 1 2 3 4 5 6 7 8非线性降维(UMAP/tSNE)
Seurat提供了多种非线性降维的方法,包括UMAP和tSNE,在低维空间上将相似的细胞放在一起,进行可视化处理。建议输入相同的PCs进行聚类分析。
# If you haven't installed UMAP, you can do so via reticulate::py_install(packages =
# 'umap-learn')
pbmc <- RunUMAP(pbmc, dims = 1:10)# note that you can set `label = TRUE` or use the LabelClusters function to help label
# individual clusters
DimPlot(pbmc, reduction = "umap")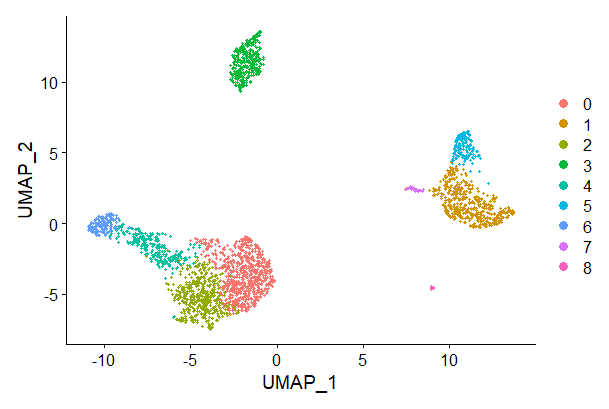
寻找差异表达基因作为聚类标记物 cluster biomarkers
默认情况下,对于某一细胞类型,Seurat会定义其相对于其他类型细胞的阳性和阴性标记物。FindMarkers()可以自动对所有细胞聚类进行此过程,也可以修改相关参数进行自定义。
min.pct 默认为0.1,指定接下来要找的标记物基因至少要在该比较组中有多少比例的细胞表达; only.pos默认为FALSE,设置为TRUE则表示只返回阳性标记物;thresh.test 参数定义两组细胞之间的差异程度。为了加快这些计算速度,我们还可以设置 max.cells.per.ident 参数,但这也会降低每个 cluster 的采样率,损失检验效能。
# find all markers of cluster 2
cluster2.markers <- FindMarkers(pbmc, ident.1 = 2, min.pct = 0.25)> head(cluster2.markers, n = 5)
p_val avg_log2FC pct.1 pct.2 p_val_adj
IL32 2.593535e-91 1.2154360 0.949 0.466 3.556774e-87
LTB 7.994465e-87 1.2828597 0.981 0.644 1.096361e-82
CD3D 3.922451e-70 0.9359210 0.922 0.433 5.379250e-66
IL7R 1.130870e-66 1.1776027 0.748 0.327 1.550876e-62
LDHB 4.082189e-65 0.8837324 0.953 0.614 5.598314e-61pct.1指该基因在第一个比较组中有多少比例的细胞表达;pct.2表示在第二个比较组中,如果没有指定第二个比较组,则表示除了第一个比较组之外的细胞中有多少比例表达。
# find all markers distinguishing cluster 5 from clusters 0 and 3
cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25)> head(cluster5.markers, n = 5)
p_val avg_log2FC pct.1 pct.2 p_val_adj
FCGR3A 2.150929e-209 4.267579 0.975 0.039 2.949784e-205
IFITM3 6.103366e-199 3.877105 0.975 0.048 8.370156e-195
CFD 8.891428e-198 3.411039 0.938 0.037 1.219370e-193
CD68 2.374425e-194 3.014535 0.926 0.035 3.256286e-190
RP11-290F20.3 9.308287e-191 2.722684 0.840 0.016 1.276538e-186# find markers for every cluster compared to all remaining cells, report only the positive ones
pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
pbmc.markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_log2FC)min.pct这个参数可以先从较低的值尝试,比如默认的0.1,后面进一步选择相对准确的marker基因时可以再进行调整。
> pbmc.markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_log2FC)
Registered S3 method overwritten by 'cli':
method from
print.boxx spatstat
# A tibble: 18 x 7
# Groups: cluster [9]
p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
<dbl> <dbl> <dbl> <dbl> <dbl> <fct> <chr>
1 1.74e-109 1.07 0.897 0.593 2.39e-105 0 LDHB
2 1.17e- 83 1.33 0.435 0.108 1.60e- 79 0 CCR7
3 0. 5.57 0.996 0.215 0. 1 S100A9
4 0. 5.48 0.975 0.121 0. 1 S100A8
5 7.99e- 87 1.28 0.981 0.644 1.10e- 82 2 LTB
6 2.61e- 59 1.24 0.424 0.111 3.58e- 55 2 AQP3
7 0. 4.31 0.936 0.041 0. 3 CD79A
8 9.48e-271 3.59 0.622 0.022 1.30e-266 3 TCL1A
9 1.17e-178 2.97 0.957 0.241 1.60e-174 4 CCL5
10 4.93e-169 3.01 0.595 0.056 6.76e-165 4 GZMK
11 3.51e-184 3.31 0.975 0.134 4.82e-180 5 FCGR3A
12 2.03e-125 3.09 1 0.315 2.78e-121 5 LST1
13 1.05e-265 4.89 0.986 0.071 1.44e-261 6 GZMB
14 6.82e-175 4.92 0.958 0.135 9.36e-171 6 GNLY
15 1.48e-220 3.87 0.812 0.011 2.03e-216 7 FCER1A
16 1.67e- 21 2.87 1 0.513 2.28e- 17 7 HLA-DPB1
17 7.73e-200 7.24 1 0.01 1.06e-195 8 PF4
18 3.68e-110 8.58 1 0.024 5.05e-106 8 PPBP我们可以用 test.use 参数设置多种检验方式。例如,用 ROC 返回任何单个分子标记的“分类效能”。
cluster1.markers <- FindMarkers(pbmc, ident.1 = 0, logfc.threshold = 0.25, test.use = "roc", only.pos = TRUE)Seurat提供的多种可视化方法:常用小提琴图VlnPlot()展示某个基因在不同类型细胞间分布差异,FeaturePlot()在tSNE和PCA图上可视化基因表达水平。可以探索一下 RidgePlot(), CellScatter(), 以及 DotPlot()等图的结果如何。
VlnPlot(pbmc, features = c("MS4A1", "CD79A"))
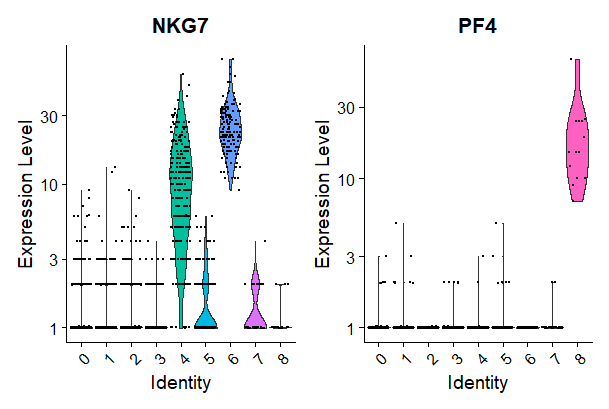
# you can plot raw counts as well
VlnPlot(pbmc, features = c("NKG7", "PF4"), slot = "counts", log = TRUE)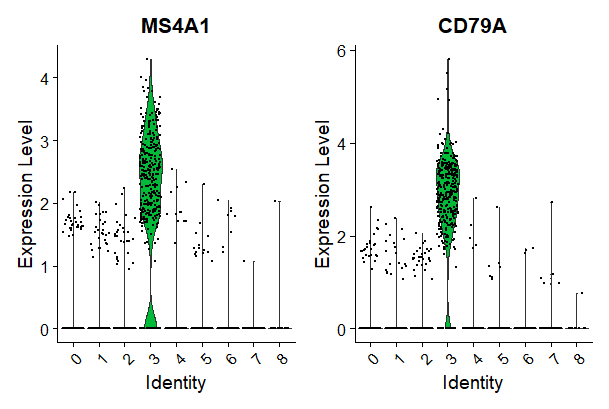
FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A"))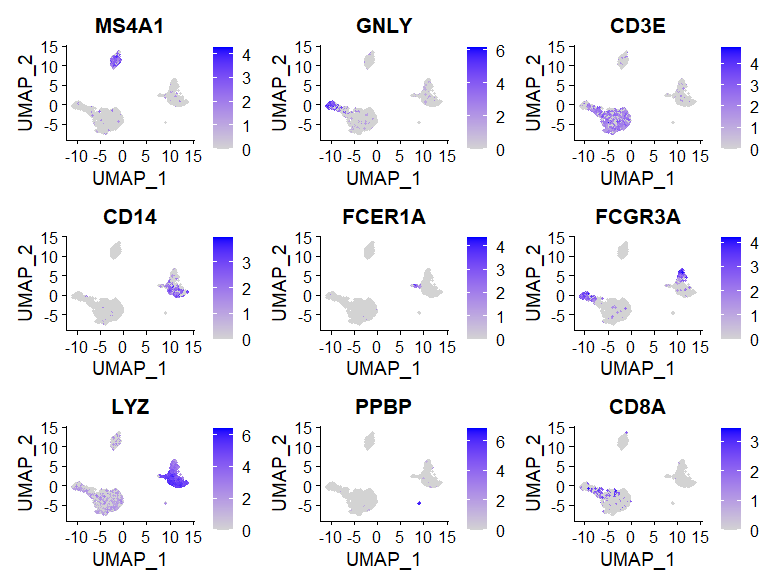
DoHeatmap() 可以为给定的细胞和基因生成一个热图。举个例子,这里绘制每个 cluster 的前10个 markers(如果小于10,则绘制所有 markers )。
top10 <- pbmc.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_log2FC)
DoHeatmap(pbmc, features = top10$gene) + NoLegend()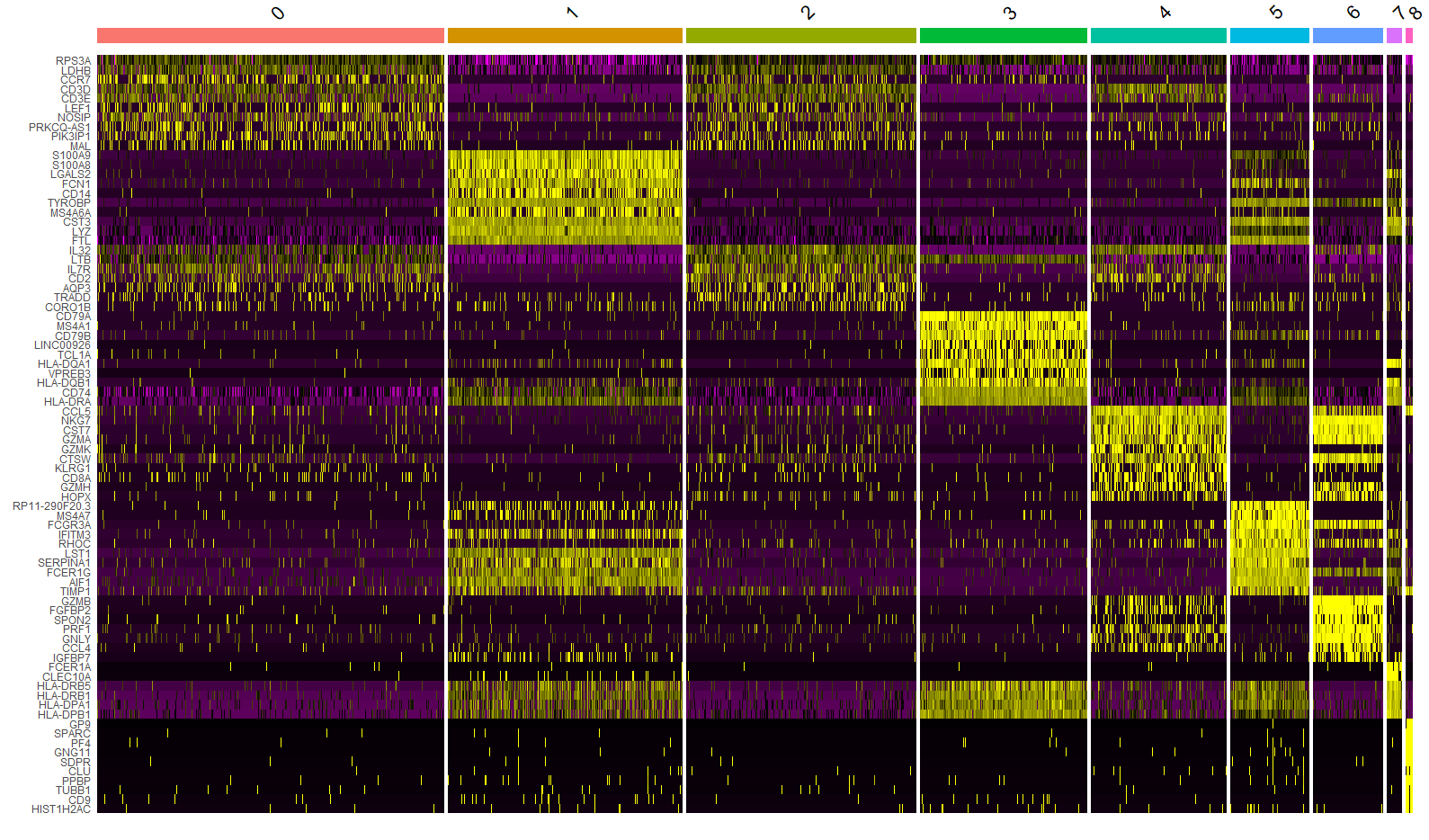
鉴定细胞类型
该示例数据可以用规范的分子标记物轻松鉴定各细胞类型。
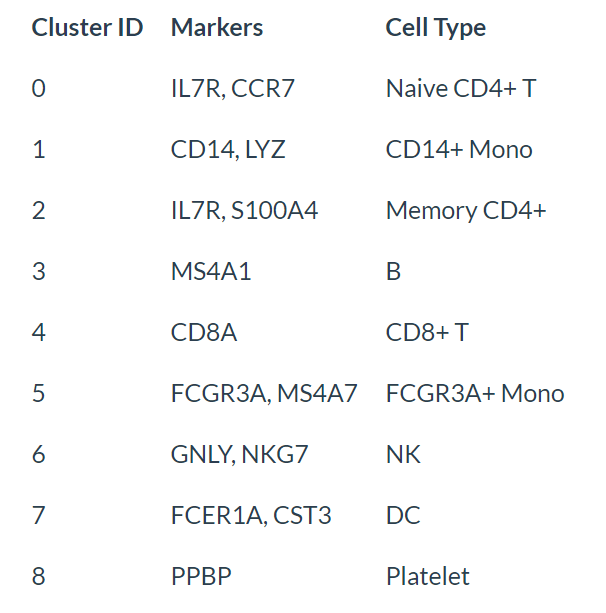
new.cluster.ids <- c("Naive CD4 T", "CD14+ Mono", "Memory CD4 T", "B", "CD8 T", "FCGR3A+ Mono",
"NK", "DC", "Platelet")
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()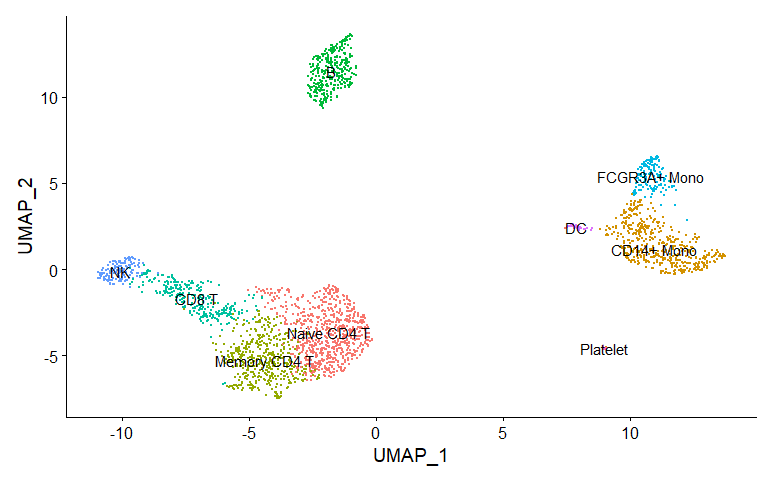
saveRDS(pbmc, file = "../output/pbmc3k_final.rds")保存为RDS文件后,以后可以直接用R读取,获得Seurat对象的全部数据。



暂无评论内容