

本文主要是翻译,用作参考,还是要看GitHub上的英文原版学习
目标
- 对于不同条件下的样本,将相同类型的细胞对齐到一起
挑战
- 对准类似的细胞类型,这样我们就不会因为样品、条件、模式或批次的不同而出现下游的聚类。
建议
- 先进行没有整合的分析,以确定是否有必要进行整合
是否有整合的必要?
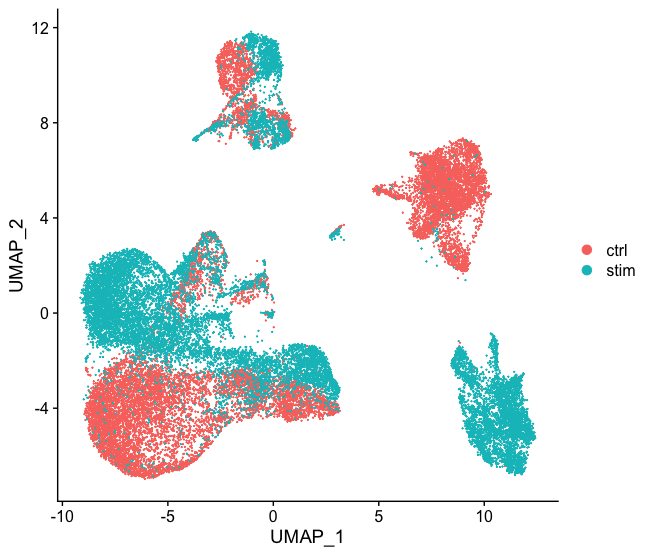
一般来说,在决定是否需要进行任何整合之前,我们总是先看看我们的聚类,而不进行整合。不要因为你认为可能有差异就总是执行整合–探索数据。如果我们在Seurat对象中对两种条件一起进行归一化,并将细胞之间的相似性可视化,我们就会看到特定条件的聚类。
通过样本间共同的高变基因进行整合
- 不同的条件 (e.g. control and stimulated)
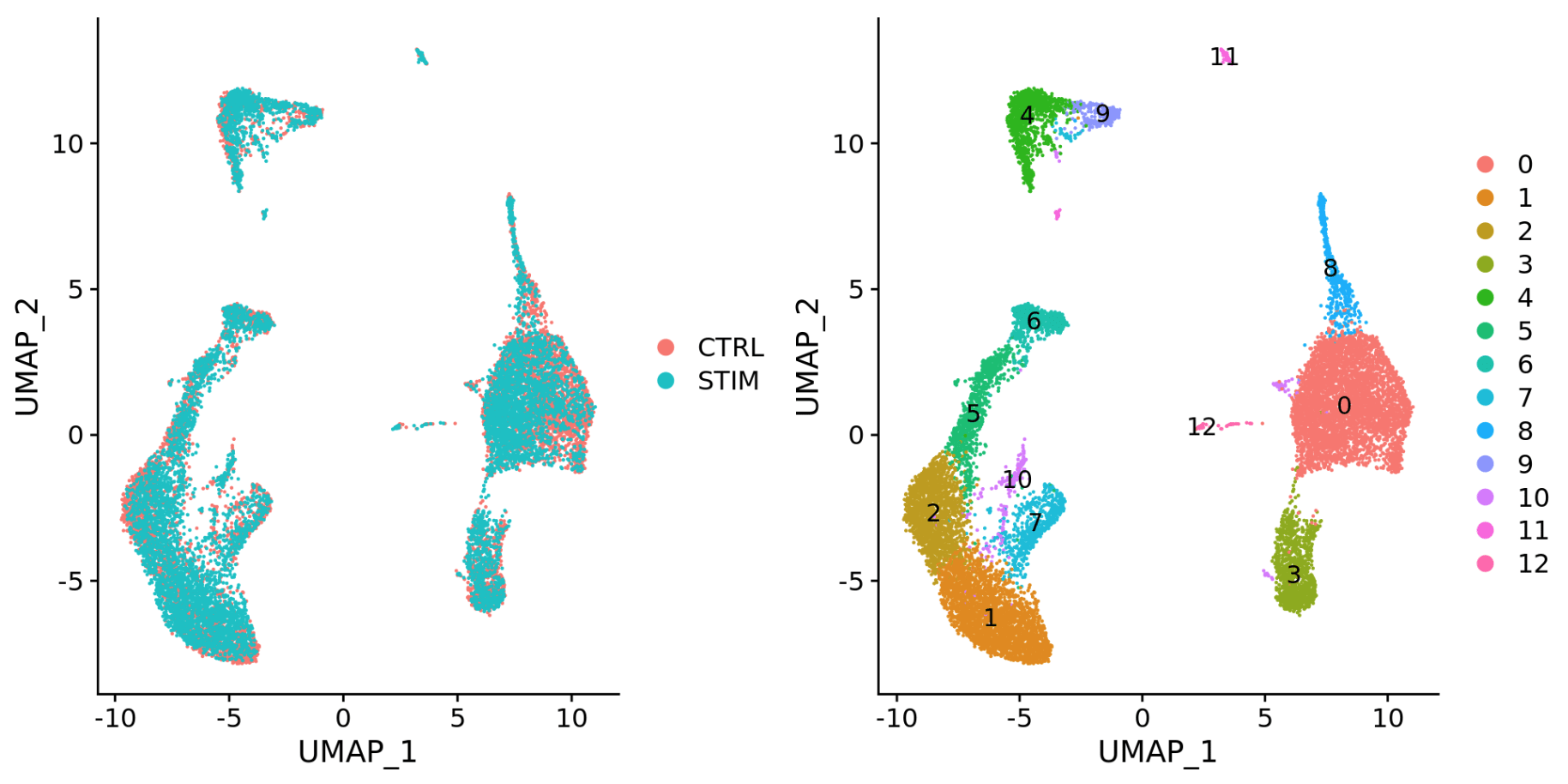
- 不同的数据集(相同样品不同的文库制备方法)
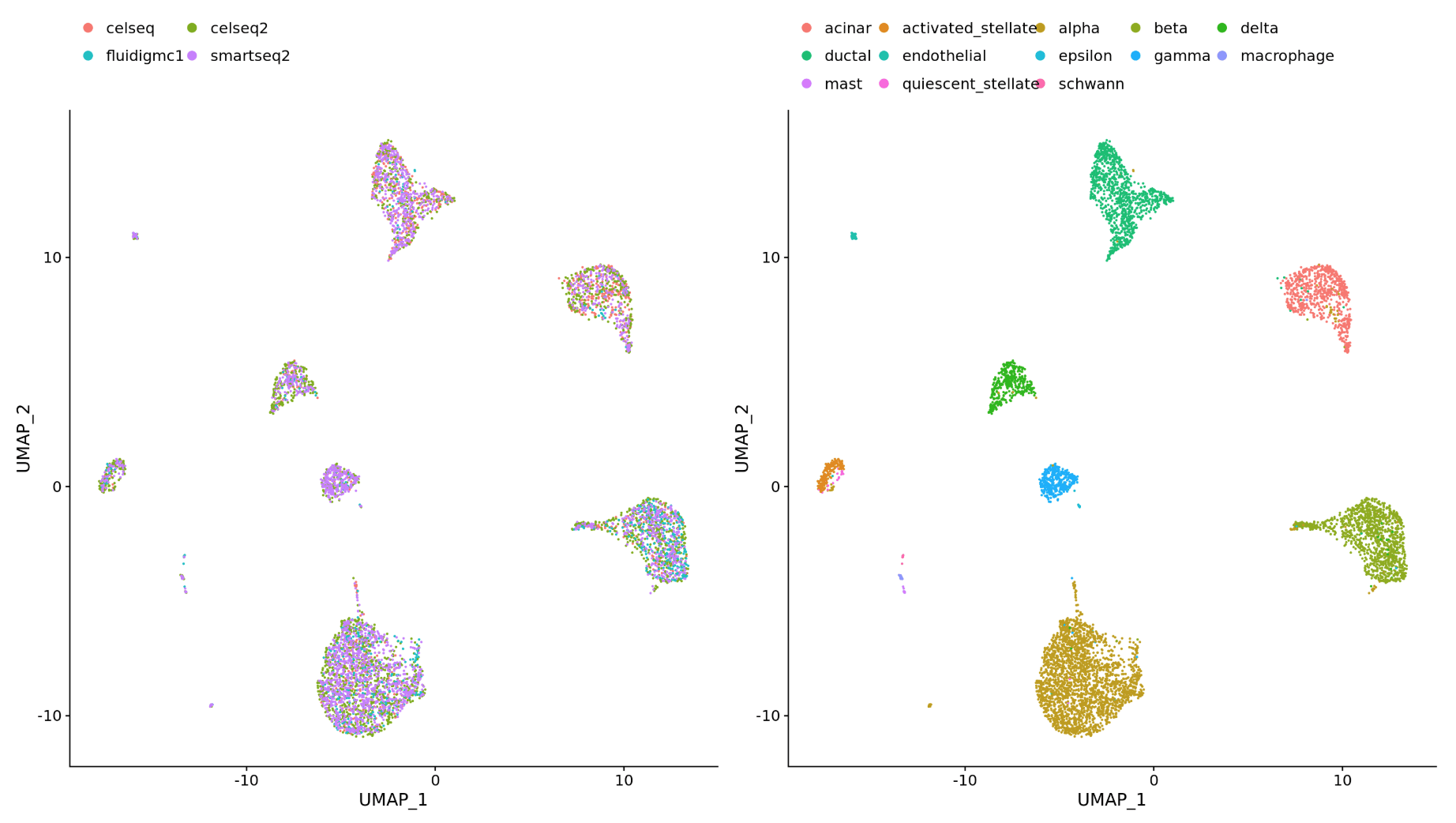
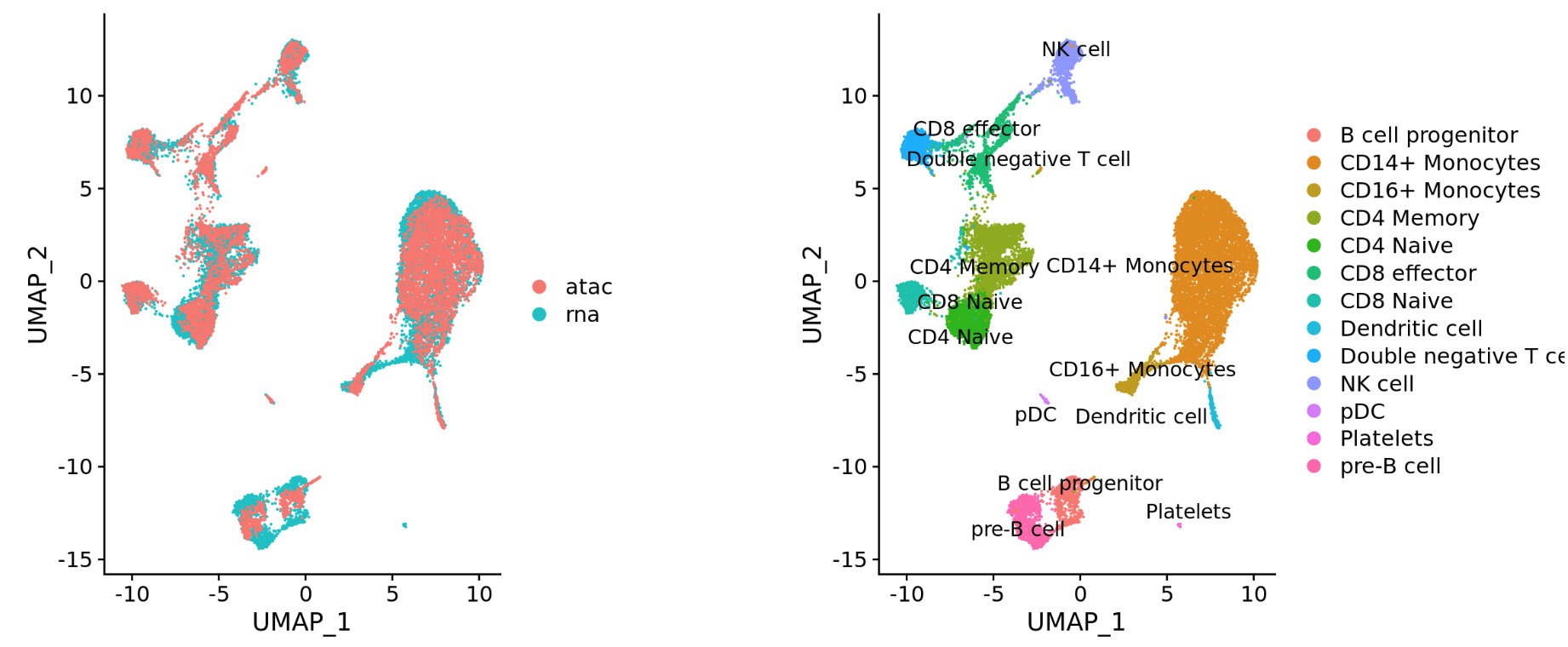
- 不同的数据类型(scRNA-seq and scATAC-seq)
- 不同的实验批次
整合是一种强大的方法,它利用这些最大变异的共享来源来识别不同条件或数据集的共享亚群[Stuart and Bulter et al. (2018)].。整合的目的是确保一个条件数据集的细胞类型与其他条件数据集的相同细胞类型一致(例如,对照组巨噬细胞与受刺激的巨噬细胞一致)。
具体来说,这种整合方法期望在各组中至少有一个单细胞子集的 "对应 "或共享生物状态。整合分析的步骤在下图中有所概述。
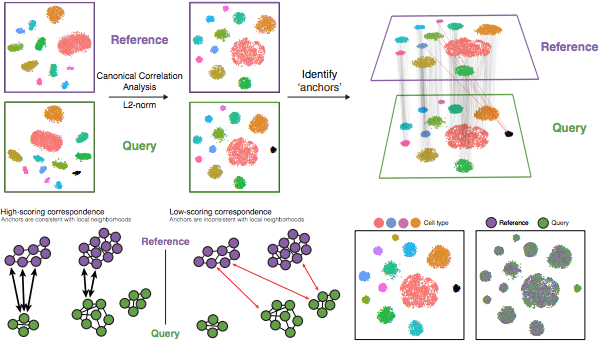
Stuart T and Butler A, et al. Comprehensive integration of single cell data, bioRxiv 2018 (https://doi.org/10.1101/460147)
整合的步骤
具体实现方法见 scRNA-seq数据整合Introduction ,标准工作流程中的整合是没有使用SCTransform的,此教程中的方法会更精细。
- 运行典型相关分析(CCA)
- 识别anchors
- 筛选anchors
- 整合
接下来进行整合:
首先,我们需要指定我们要使用由SCTransform确定的所有3000个高度变异基因进行整合。默认情况下,这个函数只选择前2000个基因。
# Select the most variable features to use for integration
integ_features <- SelectIntegrationFeatures(object.list = split_seurat,
nfeatures = 3000)现在,我们需要为整合准备SCTransform对象。
# Prepare the SCT list object for integration
split_seurat <- PrepSCTIntegration(object.list = split_seurat,
anchor.features = integ_features)现在,我们将执行CCA,查找最佳伙伴或锚点并过滤不正确的锚点。控制台中的进度条将保持为0%,但要知道它实际上正在运行。这一步运行时间会很久,取决于样本数量和选择的高变基因数量。
# Find best buddies - can take a while to run
integ_anchors <- FindIntegrationAnchors(object.list = split_seurat,
normalization.method = "SCT",
anchor.features = integ_features)接下来即可运行整合
# Integrate across conditions
seurat_integrated <- IntegrateData(anchorset = integ_anchors,
normalization.method = "SCT")UMAP降维 可视化
在整合之后,为了使整合后的数据可视化,我们可以使用降维技术,如PCA和统一面域逼近和投影(UMAP)。虽然PCA会确定所有的PC,但我们一次只能绘制两个。相比之下,UMAP将从任何数量的顶级PC中获取信息,在这个多维空间中排列单元。它将在多维空间中获取这些距离,并在二维空间中绘制它们,以保留局部和整体结构。通过这种方式,细胞之间的距离代表表达的相似性。如果你想更详细地探索UMAP,这篇文章是对UMAP理论的一个很好的介绍。
为了生成这些视觉效果,我们需要首先运行PCA和UMAP方法。让我们从PCA开始。
# Run PCA
seurat_integrated <- RunPCA(object = seurat_integrated)
# Plot PCA
PCAPlot(seurat_integrated,
split.by = "sample")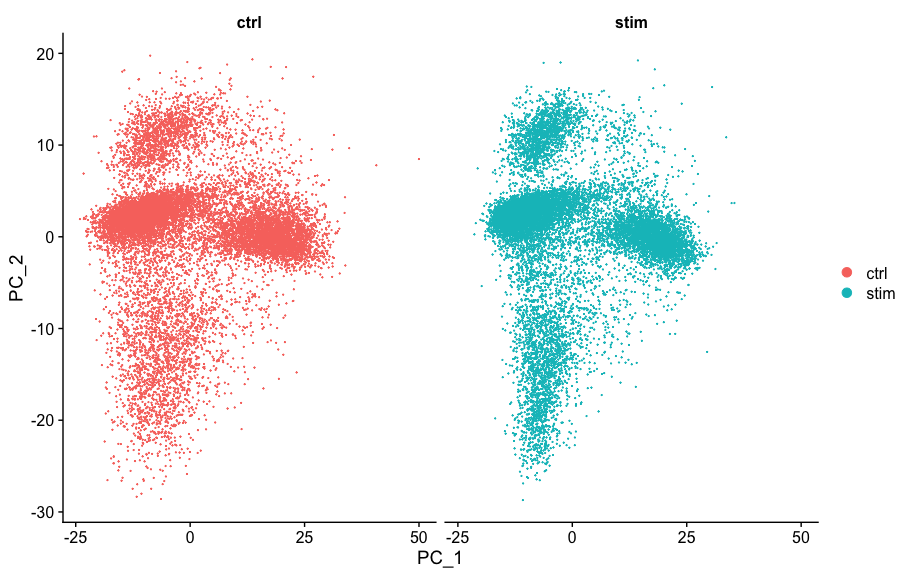
现在,我们也可以用UMAP进行可视化。让我们运行该方法并绘制。
# Run UMAP
seurat_integrated <- RunUMAP(seurat_integrated,
dims = 1:40,
reduction = "pca")
# Plot UMAP
DimPlot(seurat_integrated)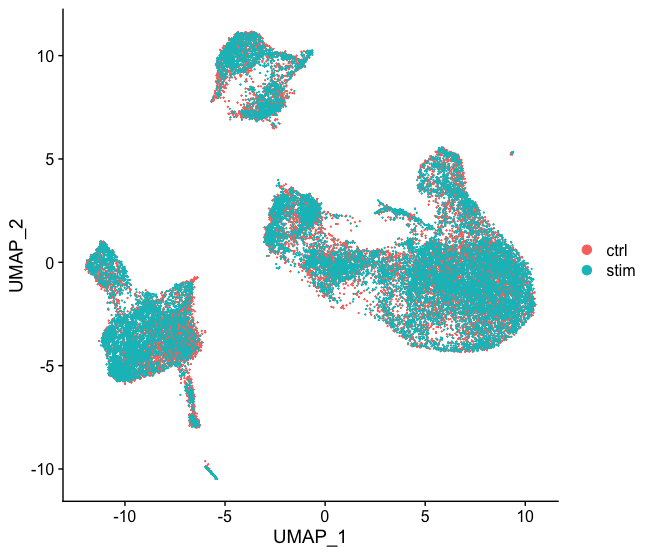
跟前面未整合的降维图比较一下,整合的效果还是很明显的。
有时候需要样本分开来比较:
# Plot UMAP split by sample
DimPlot(seurat_integrated,
split.by = "sample")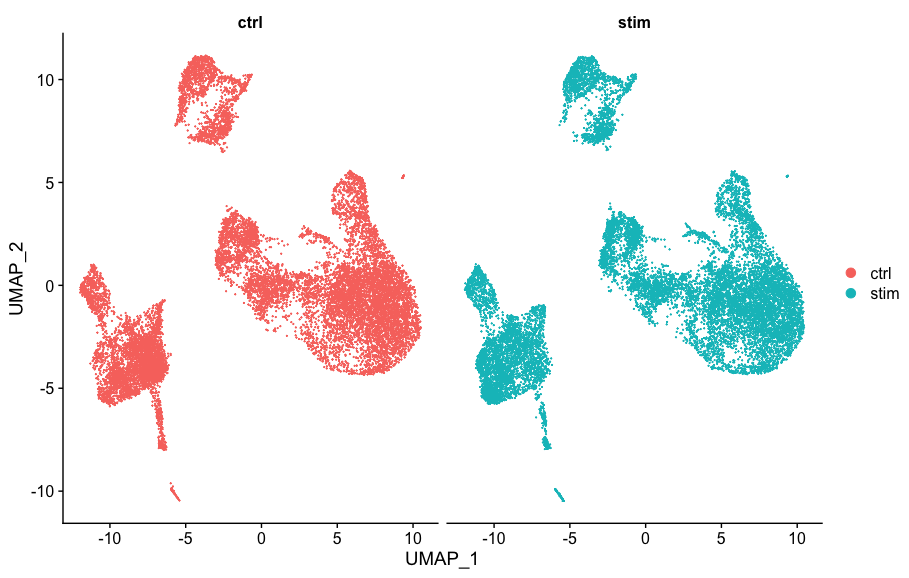
保存整合后的对象
# Save integrated seurat object
saveRDS(seurat_integrated, "results/integrated_seurat.rds")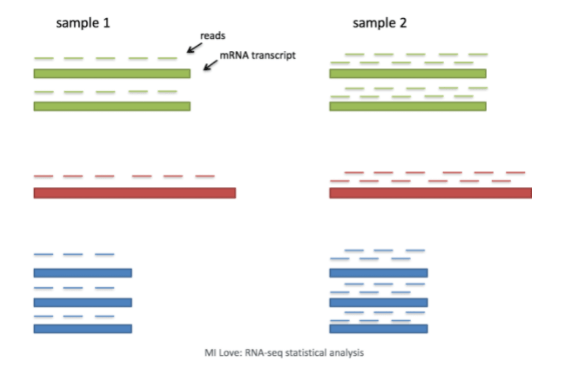

暂无评论内容