

经验证,人类样本的单细胞分析使用seurat教程的方法计算线粒体序列比例与此方法结果一致,但对于其他物种,建议使用此方法。
使用注释文件生成线粒体计数指标
我们将使用AnnotationHub,它允许加入各种各样的在线数据库和其他资源,以查询通过ensembldb提供的Ensembl注释。Ensembldb是一个软件包,可以直接从Ensembl检索数据库的注释。
安装AnnotationHub和ensembldb包
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("AnnotationHub")
BiocManager::install("ensembldb")
library(AnnotationHub)
# 阅读自带文档
browseVignettes("AnnotationHub")下载相应物种的数据集
要访问Ensembl为人类提供的各种注释,我们需要首先连接到AnnotationHub,然后指定我们感兴趣的物种和数据库。
# Connect to AnnotationHub
ah <- AnnotationHub()
# Access the Ensembl database for organism
ahDb <- query(ah,
pattern = c("Homo sapiens", "EnsDb"),
ignore.case = TRUE)接下来,我们从这个Ensembl数据库获取最新的注释文件。
我们可以先检查哪些注释版本可用:
# Check versions of databases available
ahDb %>%
mcols()由于我们想要最新的,我们将返回这个数据库的AnnotationHub ID:
# Acquire the latest annotation files
id <- ahDb %>%
mcols() %>%
rownames() %>%
tail(n = 1)最后,我们可以使用AnnotationHub连接下载相应的Ensembl数据库,其版本应该是GRCh38.92:
# Download the appropriate Ensembldb database
edb <- ah[[id]]提取基因的信息,我们可以使用Ensembldb函数genes()来返回注释的数据框:
# Extract gene-level information from database
annotations <- genes(edb,
return.type = "data.frame")提取线粒体基因ID
注释文件中信息很多,我们只需要提取我们需要的列:
# Select annotations of interest
annotations <- annotations %>%
dplyr::select(gene_id, gene_name, gene_biotype, seq_name, description, entrezid)
View(annotations)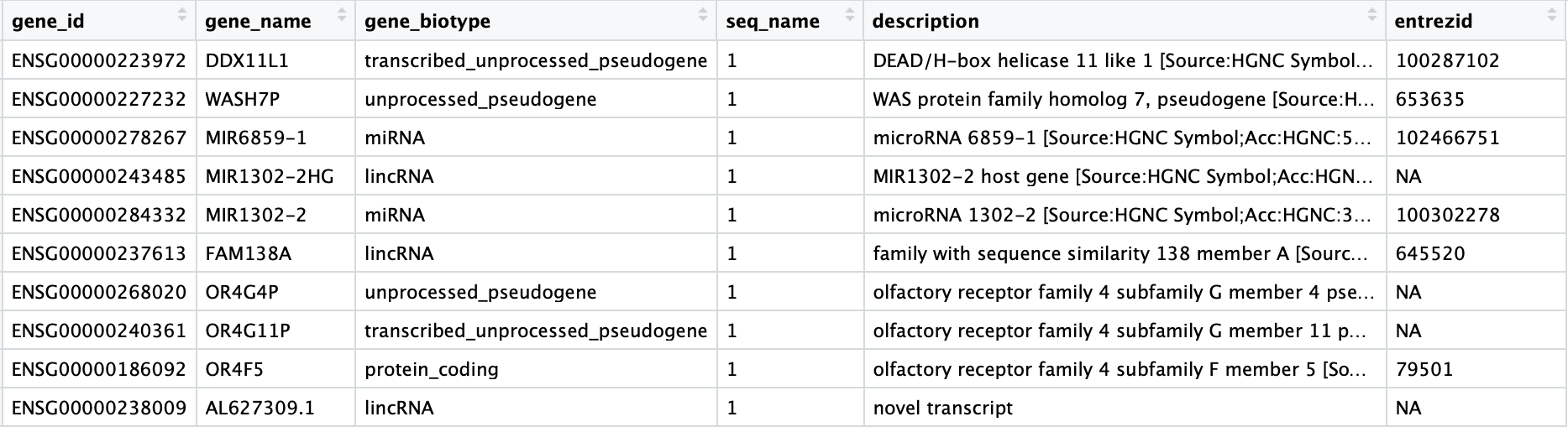
这一步可以保存一下这个注释文件,以便下次使用:
saveRDS(annotations, file = "data/annotations.rds")现在我们可以检索到与感兴趣的不同生物型相关的基因:
# Extract IDs for mitochondrial genes
mt <- annotations %>%
dplyr::filter(seq_name == "MT") %>%
dplyr::pull(gene_name)将参数添加到元数据中
# Number of UMIs assigned to mitochondrial genes
counts <- bav_merged@assays$RNA
metadata$mtUMI <- Matrix::colSums(counts[which(rownames(counts) %in% mt),], na.rm = T)
# Calculate of mitoRatio per cell
metadata$mitoRatio <- metadata$mtUMI/metadata$nUMI© 版权声明
文章版权归作者所有,转载请注明来源。
THE END



暂无评论内容