


准备工作:下载参考基因组和数据集
下载参考基因组
# 新建一个ref文件夹存放参考基因组
cd yard
mkdir ref
cd ref
# 下载human GRCH38,大约11G
wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2020-A.tar.gz
# mouse mm10 大约10G (如果有需要则下载)
curl -O https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-mm10-2020-A.tar.gz下载测序文件
# 本示例使用了10X 官网上的一个公开数据集,来自人类外周血单核细胞 (PBMC) 的1000 个 细胞 数据集,这些数据集由淋巴细胞(T 细胞、B 细胞和 NK 杀伤)和单核细胞组成。
wget https://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_1k_v3/pbmc_1k_v3_fastqs.tar
tar -xvf pbmc_1k_v3_fastqs.tar
# 解压后文件目录如下
pbmc_1k_v3_fastqs/
pbmc_1k_v3_fastqs/pbmc_1k_v3_S1_L001_R2_001.fastq.gz
pbmc_1k_v3_fastqs/pbmc_1k_v3_S1_L002_I1_001.fastq.gz
pbmc_1k_v3_fastqs/pbmc_1k_v3_S1_L001_R1_001.fastq.gz
pbmc_1k_v3_fastqs/pbmc_1k_v3_S1_L002_R1_001.fastq.gz
pbmc_1k_v3_fastqs/pbmc_1k_v3_S1_L002_R2_001.fastq.gz
pbmc_1k_v3_fastqs/pbmc_1k_v3_S1_L001_I1_001.fastq.gz分析数据
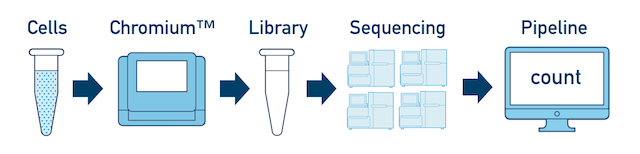
如果是一个建库样本,无论测多少次,都可以直接用cellranger count来做分析。但如果是不同的建库样本,想要放在一起分析就需要:先用cellranger count分别定量,然后用cellranger aggr合并样本。但是一般只用到cellranger count,整合的过程使用下游分析软件,如seurat
我自己目前只用到cellranger count,所以只记录一下这个功能如何使用。
cellranger count --id=stenosis04 --fastqs=./SRR10134385 --sample=SRR10134385 --transcriptome=/home/vamond/yard/ref/refdata-gex-GRCh38-2020-A定量分析
本教程中的数据是大约1000个细胞,使用个人电脑内存至少16G,否则有可能跑不出来。
实际上真正分析项目数据的时候16G是远远不够的,官方建议最低标准为64G,128G以上更佳。
我们需要用到cellranger count命令来进行定量分析,可以先看命令的帮助文档,帮助我们理解其参数的含义。
cellranger count --help我使用的是官方教程的参数:
cellranger count --id=run_count_1kpbmcs \
--fastqs=/mnt/home/user.name/yard/run_cellranger_count/pbmc_1k_v3_fastqs \
--sample=pbmc_1k_v3 \
--transcriptome=/home/vamond/yard/ref/refdata-gex-GRCh38-2020-A了解一下参数含义:
—id 写项目名称,cellranger会自动生成这个文件夹
—fastqs 填写fastq文件的目录
—sample 即样本的名称
—transcriptome 填写参考序列的路径
其他可能常用的参数:
–no-bam 不需要bam文件
–nosecondary 不需要它分析聚类和细胞分群
–disable-ui 不需要web展示
结果展示
这 1000 个细胞,我的电脑是8核16线程的处理器,大概几十分钟跑完吧,总体来说还是比较快的。输出结果中我们主要看 outs 目录下面的文件:
# 列出所有文件
ls -1 run_count_1kpbmcs/outs
# outs文件夹下的文件目录一般是这样的
analysis
cloupe.cloupe
filtered_feature_bc_matrix
filtered_feature_bc_matrix.h5
metrics_summary.csv
molecule_info.h5
possorted_genome_bam.bam
possorted_genome_bam.bam.bai
raw_feature_bc_matrix
raw_feature_bc_matrix.h5
web_summary.htmlweb_summary.html这个页面就是cellrager自动生成的报告,里面包括了他自动聚类的结果。filtered_feature_bc_matrix 和 raw_feature_bc_matrix 这两个文件夹就是可以用作下游分析的表达矩阵数据了,两者的区别在于一个是经过cellranger筛选识别去掉了不是细胞的barcodes,另一个是所有barcodes的数据。
补充
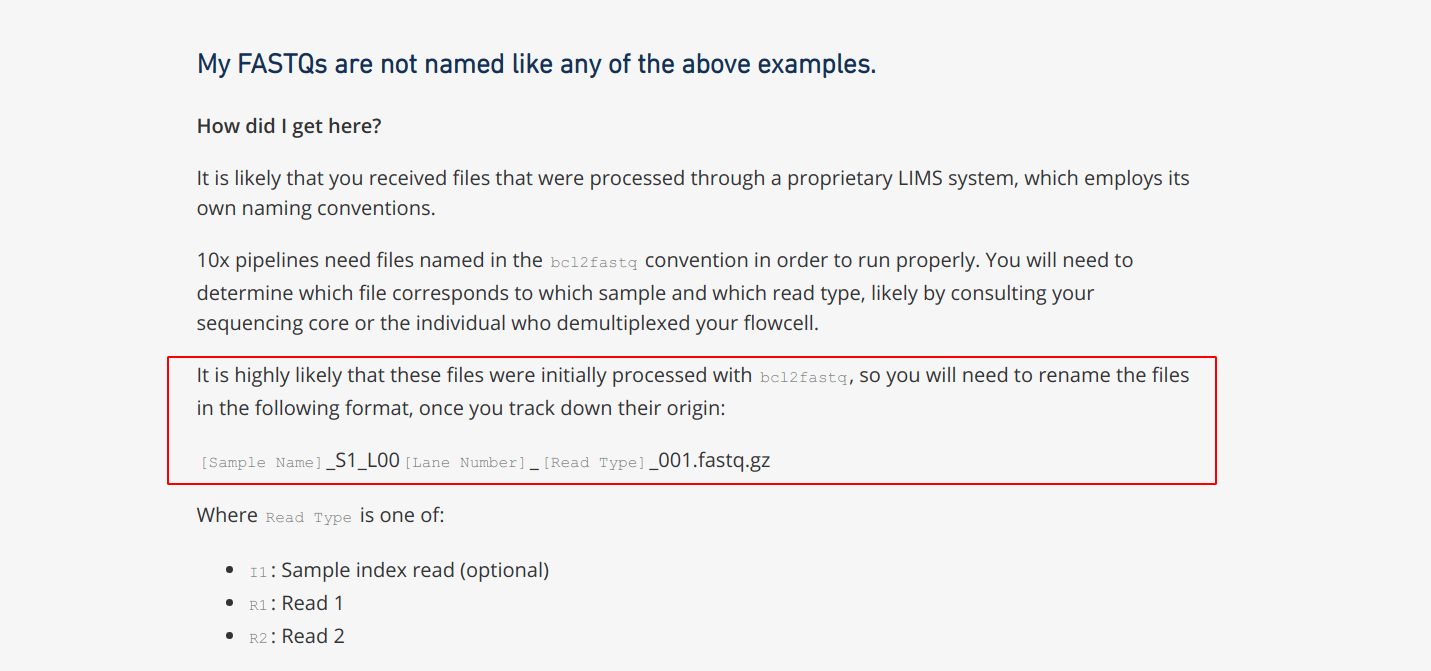
教程中提供的fastq数据都是命名好的能够直接作为cellranger count 的输入文件,但是如果是我们自己在数据库中下载的文件,如果是rsa文件,需要先转换成fastq;如果是下载的fastq文件,通常也是需要自己根据cellranger的要求进行重命名后才能作为输入的。


暂无评论内容