

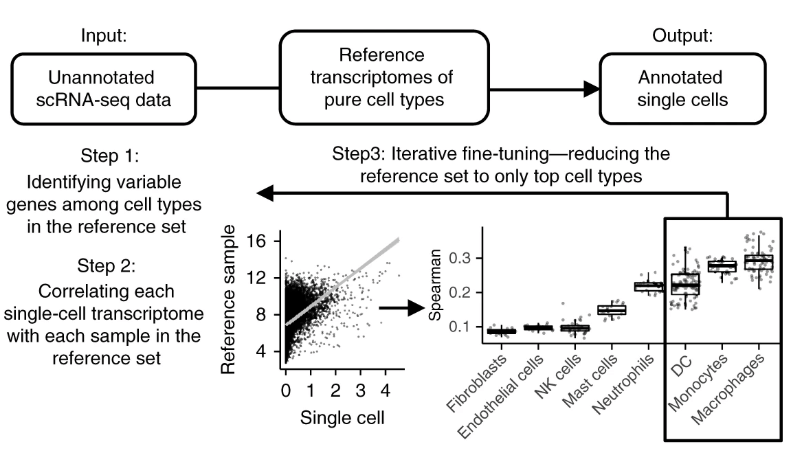
SingleR是用于单细胞RNA测序(scRNAseq)数据的自动注释方法(Aran et al.2019)。给定具有已知标签的样本(单细胞或RNAseq)参考数据集,它将基于与参考数据的相似性标记测试数据集中的新细胞。具体来说,对于每个测试单元:
- 计算其表达谱与每个参考样品的表达谱之间的Spearman相关性。
- 将每个标签的分数定义为相关性分布的fixed quantile(默认为0.8)。
- 对所有标签重复此操作,然后将得分最高的标签作为此细胞的注释。
- 选择性执行微调
安装
BiocManager::install("SingleR")
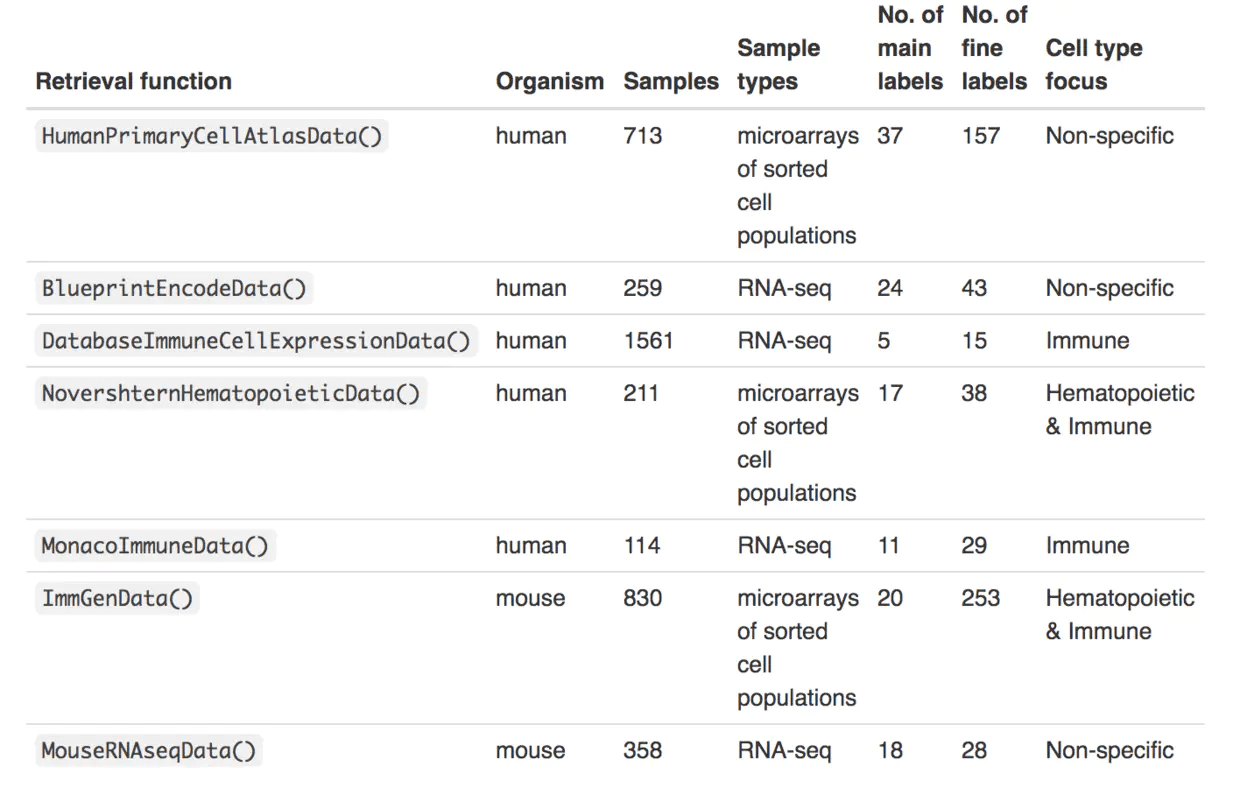
BiocManager::install("celldex")SingleR通过专用的数据检索功能提供了多个参考数据集(主要来自大量RNA-seq或微阵列数据)。例如,我们使用HumanPrimaryCellAtlasData()函数从人类原代细胞图集获得参考数据,该函数返回一个SummarizedExperiment对象,该对象包含带有样本级标签的对数表达值矩阵。
singleR的自带参考数据集保存在celldex包中,其中五个人类参考数据和两个小鼠参考数据:
使用
library(SingleR)
ref <- HumanPrimaryCellAtlasData()
ref
pred.scRNA <- SingleR(test = scRNA@assays$RNA@data, ref = ref,labels = ref$label.main, clusters = scRNA@active.ident, fine.tune = TRUE, BPPARAM = MulticoreParam(40))
#鉴定各聚类的细胞类型,可选择method 参数为“single”,返回每个细胞的鉴定结果。clusters参数指定的各聚类的名称
pred.scRNA$pruned.labels
#查看注释准确性
pdf(file="10.celllabel.pdf",width=10,height=10)
plotScoreHeatmap(pred.scRNA, clusters=pred.scRNA@rownames, fontsize.row = 9,show_colnames = T)
dev.off()
#绘制带cell label的tsne和umap图
new.cluster.ids <- pred.scRNA$pruned.labels
names(new.cluster.ids) <- levels(scRNA)
levels(scRNA)
# 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
scRNA <- RenameIdents(scRNA,new.cluster.ids)
levels(scRNA)
pdf(file="10.TSNE_lable.pdf",width=6.5,height=6)
TSNEPlot(object = scRNA1, pt.size = 0.5, label = TRUE)
dev.off()
pdf(file="10.uMAP_label.pdf",width=6.5,height=6)
DimPlot(scRNA1, reduction = "umap",pt.size=0.5,label = TRUE)
dev.off()© 版权声明
文章版权归作者所有,转载请注明来源。
THE END


暂无评论内容