

Harmony方法在2019年发表在上面nature methods,
harmony算法与其他整合算法相比的优势:
- 整合数据的同时对稀有细胞的敏感性依然很好;
- 省内存;
- 适合于更复杂的单细胞分析实验设计,可以比较来自不同供体,组织和技术平台的细胞。
基本原理
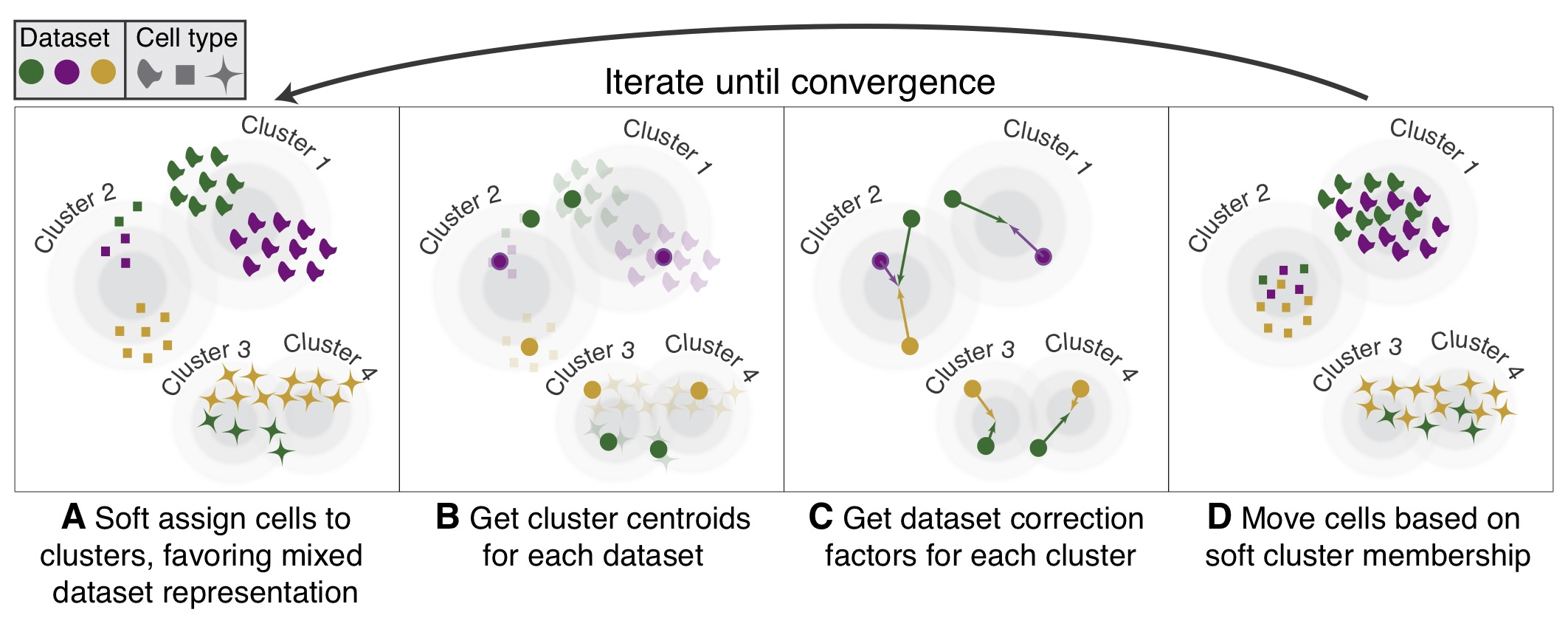
我们用不同颜色表示不同数据集,用形状表示不同的细胞类型。首先,Harmony应用主成分分析将转录组表达谱嵌入到低维空间中,然后应用迭代过程去除数据集特有的影响。
(A)Harmony概率性地将细胞分配给cluster,从而使每个cluster内数据集的多样性最大化。
(B)Harmony计算每个cluster的所有数据集的全局中心,以及特定数据集的中心。
(C)在每个cluster中,Harmony基于中心为每个数据集计算校正因子。
(D)最后,Harmony使用基于C的特定于细胞的因子校正每个细胞。由于Harmony使用软聚类,因此可以通过多个因子的线性组合对其A中进行的软聚类分配进行线性校正,来修正每个单细胞。重复步骤A到D,直到收敛为止。聚类分配和数据集之间的依赖性随着每一轮的减少而减小。
在R中安装harmony
library(devtools)
install_github("immunogenomics/harmony")使用Seurat进行PCA
library(Seurat)
library(tidyverse)
library(dplyr)
library(patchwork)
library(harmony)harmony要求输入的对象必须含有PCA坐标。运行NormalizeData(), FindVariableFeatures(), ScaleData(), RunPCA()后,得到相应的seurat对象。具体操作见 Seurat基本分析流程
使用harmony进行整合
在seurat流程中使用harmony非常方便,只需要注意以下两点:
- 运行harmony的函数为
RunHarmony() - 在下游分析中,降维方式选择
harmony而不是默认的pca
比如,需要运行UMAP,则使用以下代码:
seuratObj <- RunHarmony(seuratObj, "dataset")
seuratObj <- RunUMAP(seuratObj, reduction = "harmony")在分析流程上,按照seurat标准流程进行即可。
© 版权声明
文章版权归作者所有,转载请注明来源。
THE END


暂无评论内容