

本篇教程将会帮助你理解如何获得单细胞RNA测序实验中的数据。
基因表达定量完成之后,我们需要将这些数据导入R中,以生成可用于执行质控的矩阵。在本课中,我们将讨论计数数据可被导入的格式,以及如何将其读入R中,以便我们可以转到工作流程中的质控步骤。我们还会讨论我们将使用的数据集和相关的元数据(metadata)。
探索示例数据集
在本次教程上,我们使用的单细胞RNA测序数据集是Kang et al, 2017研究的一部分。本文提出了一种利用遗传变异(eQTL)来确定含有单个细胞(singlet)每个液滴的遗传特性,以及从不同个体中识别含有两个细胞的液滴(doublets)的计算算法。 用于测试其算法的数据由8名狼疮患者的外周血单个核细胞(PBMCs)组成,分为对照组和干扰素β治疗(刺激)组。
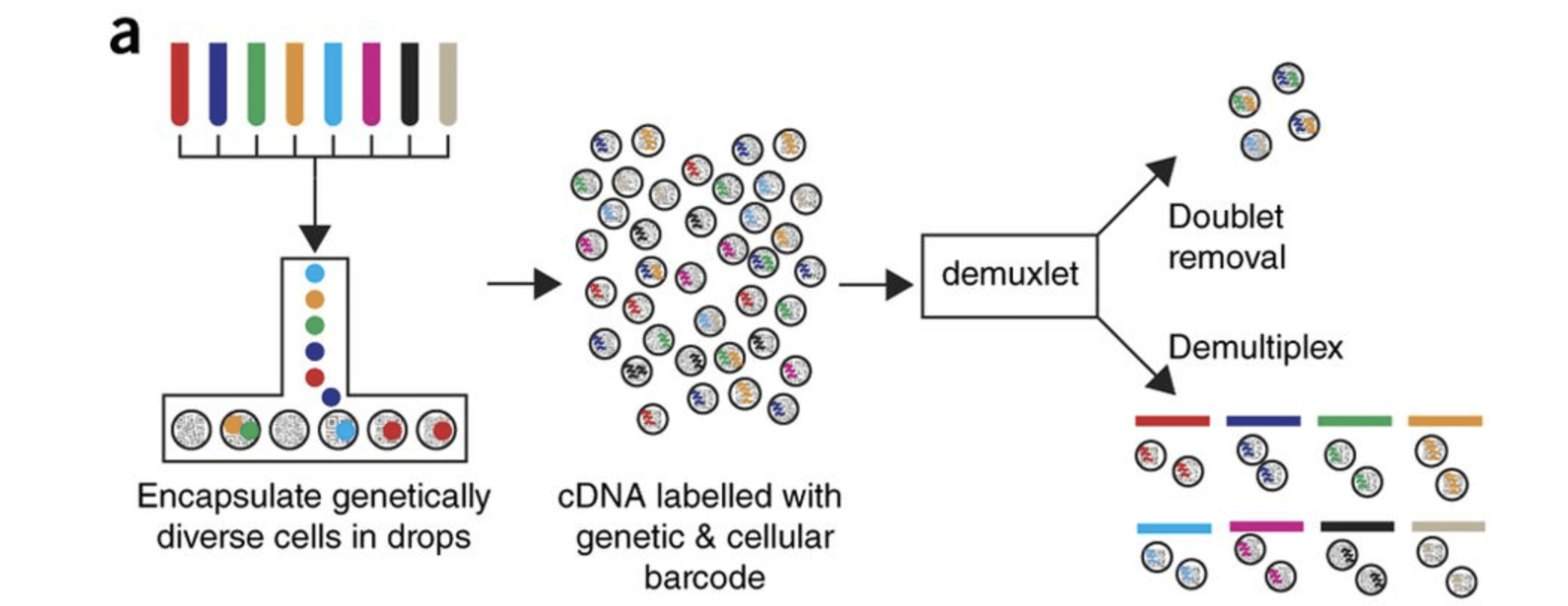
原始数据
该数据集在GEO (GSE96583)上可用, 但是可用的计数矩阵缺少线粒体序列, 因此我们从SRA (SRP102802)下载了BAM文件。这些BAM文件转换回FASTQ文件,然后通过运行 Cell Ranger来获得我们将要使用的计数(count)数据。
注:seurat官方教程也使用了此数据集。
元数据(Metadata)
除了原始数据之外,我们还需要收集有关数据的信息;这就是所谓的元数据(metadata)。人们常常会急于处理测序数据,但如果我们对这些数据来源的样本一无所知,那就没有什么意义了。
我们数据集的一些相关元数据如下:
- 这些文库是用10X Genomics第二版化学方法制备的
- 样品在illumina NextSeq 500测序仪上进行测序
- 将8例狼疮患者的PBMCs分成两组
- 一组PBMCs加入100U/ml重组IFN-β刺激,持续6小时。
- 第二组PBMCs不经任何处理
- 6小时后,将每种条件下的8个样本汇集到两个最终组中(受刺激细胞和对照细胞)。我们将处理这两个集合样本。
- 对照组和受刺激的混合样本分别捕获了12138和12167个细胞(去除双重液滴后)。
- 因为样本是PBMC,我们期望划分出免疫细胞,比如:
- B细胞
- T细胞
- NK细胞
- 单核细胞
- 巨噬细胞
- 似巨核细胞
建议您在执行质控之前对希望在数据集中看到的细胞类型有一定的了解。这将告诉你,你是否有任何细胞类型的复杂性较低(几个基因产生许多转录本)或细胞线粒体表达水平较高。这将使我们能够在分析工作流程中考虑这些生物因素。
上述细胞类型均不具有低复杂性或高线粒体含量。
设置R环境
在涉及大量数据的研究中,最重要的部分之一是如何最好地管理数据。我们往往倾向于优先考虑分析,但数据管理的许多重要方面常常被忽视。
数据管理的一个重要方面是组织(organization)。对于您需要处理和分析数据的每个实验,最好通过创建有计划的存储空间(directory structure)来组织。我们将会在我们的单细胞分析中那样做。
首先,下载并安装Rstudio。
打开Rstudio,点击右上角的Project按钮,并创建一个名为single_cell_rnaseq的新的R项目。
然后,创建以下目录:
single_cell_rnaseq/
├── data
├── results
└── figures下载数据
右键单击下面的链接,将每个示例的输出文件夹从Cell Ranger下载到data文件夹中:
现在,让我们解压缩刚才下载的两个压缩文件夹,这样我们就可以在RStudio中看到它们的内容。
如果下载速度过慢,可尝试从蓝奏云下载:
链接: https://vamond.lanzous.com/b02621f8f 密码: a0jo
创建新脚本
接下来,点击Rstudio左上角的+号,新建一个R script文件,并在开头加上一些注释,以说明此文件将包含的内容:
# February 2020
# HBC single-cell RNA-seq workshop
# Single-cell RNA-seq analysis - QC将R script另存为quality_control.R。你的工作目录应该如下所示:
安装R包
安装分析需要的各种R包和依赖,主要包括Seurat。
Seurat是一个用于单细胞基因组学的R工具集,由纽约基因组中心的Satija实验室开发并维护。
install.packages("RCurl")
install.packages("BiocManager")
BiocManager::install("SingleCellExperiment")
BiocManager::install("tidyverse")
BiocManager::install('multtest')
install.packages('Seurat')
install.packages('hdf5r')载入R包
现在,我们可以载入必要的程序库。
# 载入R包
library(SingleCellExperiment)
library(Seurat)
library(tidyverse)
library(Matrix)
library(scales)
library(cowplot)
library(RCurl)导入单细胞RNA测序计数数据
不管处理单细胞RNA测序数据所使用的技术或流程如何,输出通常都是相同的。也就是说,对于每个单独的样本,你将拥有以下三个文件:
- 一个带有细胞ID (cell IDs)的文件,代表所有被定量的细胞
- 一个带有基因ID (gene IDs)的文件,代表所有被定量的基因
- 一个含有每个细胞每个基因的计数矩阵 (matrix of counts)
我们可以通过点击data/ctrl_raw_feature_bc_matrix文件夹来浏览这些文件:
1. barcodes.tsv
这是一个文本文件,其中包含该样本的所有细胞条码。条码按矩阵文件中展现的数据顺序列出(即这些是列名)。

2. features.tsv
这是一个文本文件,其中包含定量基因的标识符。标识符的来源可以根据定量方法中使用的参照 (reference)(即Ensembl、NCBI、UCSC)而有所不同,但大多数情况下,这些都是官方基因标志 (official gene symbol)。这些基因的顺序与矩阵文件中行的顺序相对应(即这些是行名)。

3. matrix.mtx
这是一个包含计数值矩阵的文本文件。行与上面的基因ID相关联,列与细胞条码相对应。注意这个矩阵中有很多零值。
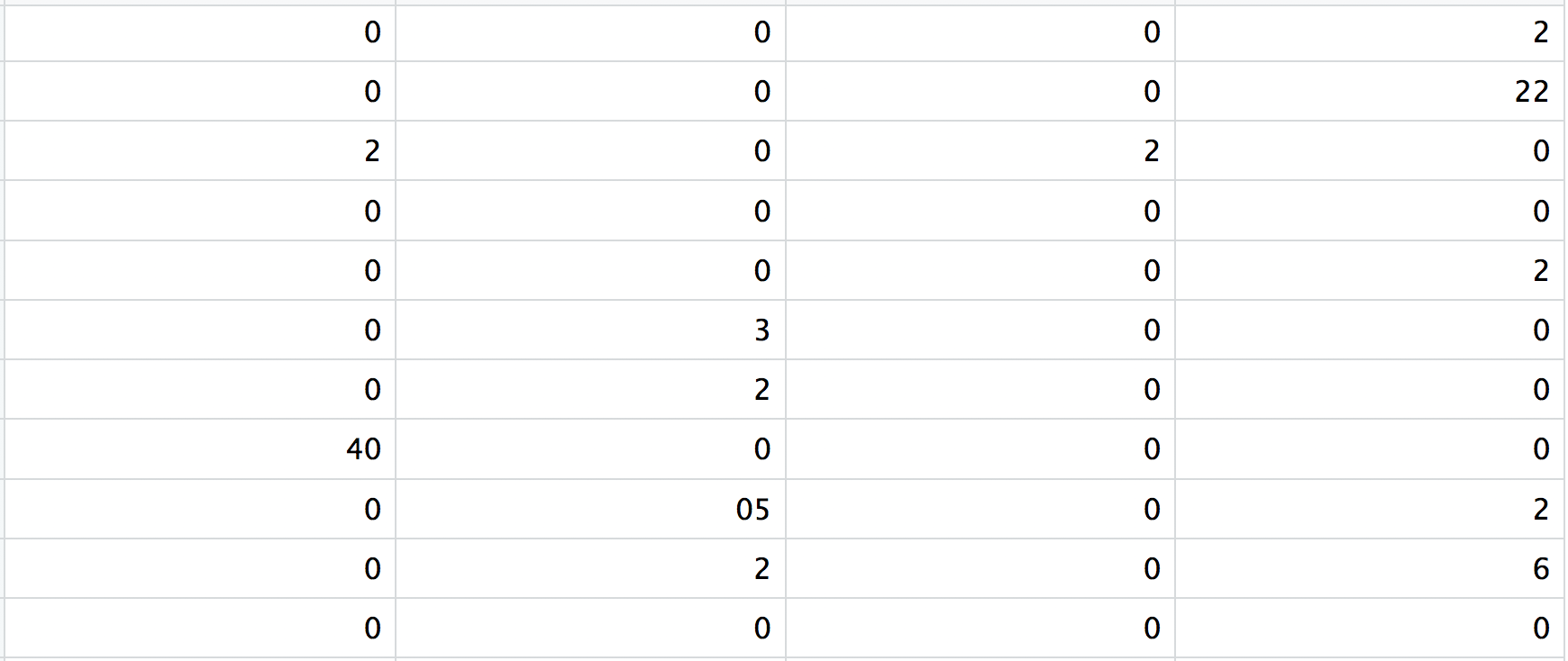
将这些数据加载到R中需要使用一些函数 (functions),这些函数允许我们有效率地将这三个文件合并到一个计数矩阵中。然而,我们不会去创建一个规则的矩阵数据结构,而是使用这些函数创建一个稀疏矩阵 (sparse matrix) ,以提高用于处理这些数据的计算性能。
读取数据的不同方法包括:
readMM():此函数来自Matrix包,将标准矩阵转换为稀疏矩阵。features.tsv文件和barcodes.tsv必须首先单独加载到R中,然后将它们组合在一起。Read10X():此函数来自Seurat包,将使用Cell Ranger输出目录作为输入。这样就不需要加载单个文件了,相反,该函数将为你加载并将它们组合成稀疏矩阵。我们将使用此函数加载我们的数据!
使用Read10X()读取单个样本数据
使用10X数据及其专有软件Cell Ranger时,你将始终有一个outs目录。在此目录中,你将找到许多不同的文件,包括:
web_summary.html:含有不同质量控制指标的报告,包括映射指标 (mapping metrix)、筛选阈值 (filtering thresholds)、筛选后的估计细胞数,以及筛选后每个细胞的读取数和基因数信息。- BAM alignment files:用于可视化映射读取和重新创建FASTQ文件的文件
filtered_feature_bc_matrix:使用Cell Ranger筛选后的数据,其中的文件可以构建完整的表达矩阵raw_feature_bc_matrix:包含使用原始未过滤数据构造计数矩阵所需的所有文件的文件夹
我们主要对raw_feature_bc_matrix感兴趣,因为我们希望在考虑实验/生物系统的生物学特性的同时,执行我们自己的质控和过滤操作。
如果我们只有一个样本,我们可以生成一个计数矩阵,然后创建一个Seurat对象:
# 如何读入单个样本的10X数据(输出为一个稀疏矩阵)
ctrl_counts <- Read10X(data.dir = "data/ctrl_raw_feature_bc_matrix")
# 如何将计数矩阵转化为一个Seurat对象(输出为一个Seurat对象)
ctrl <- CreateSeuratObject(counts = ctrl_counts,
min.features = 100)注:参数min.features规定了每个细胞需要被检测到的最少基因数目。这个参数将会筛掉低质量的细胞,这些油滴可能只封装了随机的细胞条码,但是其中没有任何细胞。通常情况下,检测到的基因少于100个的细胞不被考虑用于分析。
当使用Read10X()函数读取数据时,Seurat会自动为每个细胞创建一些元数据。此信息存储在Seurat对象内的meta.data插槽(slot)中(请参阅下面的注释中的更多内容)。
# 查看元数据
head(ctrl@meta.data)元数据的列代表着什么?
orig.ident:通常包含所知的样品名,默认为“SeuratProject“nCount_RNA:每个细胞的UMI数目nFeature_RNA:每个细胞所检测到的基因数目
使用for循环读入多个样本数据
实际上,在使用前面讨论的两个函数(Read10X()或ReadMM())时,你可能会需要读入多个样本的数据。因此,为了更有效率地把数据导入到R中,我们可以使用for循环,它将对给定的每个输入执行一系列命令。
在R中,它具有以下结构/语法:
## 不要运行
for (variable in input){
command1
command2
command3
}我们现在所使用的for循环将会遍历 (iterate) 两个样本“文件”,并对每个样本执行两个命令——(1)使用Read10X()读取计数数据和(2)使用CreateSeuratObject()从读取的数据中创建Seurat对象:
# 给每个样本创建单独的Seurat对象
for (file in c("ctrl_raw_feature_bc_matrix", "stim_raw_feature_bc_matrix")){
seurat_data <- Read10X(data.dir = paste0("data/", file))
seurat_obj <- CreateSeuratObject(counts = seurat_data, min.features = 100, project = file)
assign(file, seurat_obj)
}-
我们可以将
for循环拆解开来描述每一行代码的含义第一步:确定输入对于这个数据集来说,我们有两个需要创建Seurat对象的样本:
ctrl_raw_feature_bc_matrixstim_raw_feature_bc_matrix
我们可以在for循环的输入部分使用
c()将这些将这些样本指定为一个向量 (vector) 的元素。我们将这些赋值 (assign) 给一个变量 (variable),这样我们可以随意改变那个变量的命名。在本例中,我们调用了变量file。在执行以上循环的过程中,
file将会首次包含值ctrl_raw_feature_bc_matrix,并一直运行到assign()命令。接下来,它将包含值stimulations_raw_feature_bc_matrix,并再次运行所有命令。如果有15个而不是2个文件夹作为输入,则以上代码将运行15次。第二步:读取输入数据我们将会添加一行,使用
Read10X()读入数据来继续我们的for循环:- 在这里,我们需要指定文件的路径,因此我们将使用
paste0函数将data/目录前置到样本文件夹名称。
第三步:从10X计数数据中创建Seurat对象现在,我们可以使用
CreateSeuratObject()函数创建Seurat对象,并添加参数project来指定样品名称。第四步:根据样本名将Seurat对象赋值给一个新变量最后一个命令将创建的Seurat对象 (
seurat_obj) 赋值给一个新变量。这样,当我们迭代并转到input中的下一个样本时,我们不会覆盖在上一次迭代中创建的Seurat对象:Tips:运行
rm(list = c('seurat_data', 'seurat_obj'))删除两个临时文件节省内存
现在,我们已经创建了这两个对象,可以查看元数据以了解其基本信息:
# 查看新Seurat对象的元数据
head(ctrl_raw_feature_bc_matrix@meta.data)
head(stim_raw_feature_bc_matrix@meta.data)接下来,我们需要将这些对象合并成一个Seurat对象。这样会方便我们同时给每个样品运行质控步骤,并且确保我们可以更容易地比较所有样本的数据质量。
我们可以使用Seurat包中的merge()函数执行这个操作:
# 创建一个合并的Seurat对象
merged_seurat <- merge(x = ctrl_raw_feature_bc_matrix,
y = stim_raw_feature_bc_matrix,
add.cell.id = c("ctrl", "stim"))因为同一个barcode可以用于不同的样本,所以我们使用add.cell.id参数将样品特异的前缀添加到每个细胞ID。如果查看合并对象的元数据,我们能够看到行名的前缀:
# 查看合并对象是否有对应的样本前缀
head(merged_seurat@meta.data)
tail(merged_seurat@meta.data)

暂无评论内容