


对于研究比较多的分子,我们需要引物的时候可以很方便的在文献中找到别人用过的,或者在primerbank中查到相应的引物序列,然后通过比对和预实验验证选择合适的引物。但有时候我们研究的分子引物不太好找,或者我们的实验模型的物种不是人或小鼠,这时候掌握引物设计方法或许能提升不少效率。
使用的工具
qPCR引物设计的工具有很多,比如Primer-BLAST,Primer3,5、6,Primer Express,Beacon Designer,Array Designer,MRPrimerW,Genome Compiler等等,这其中有在线工具,也有桌面版软件,本文主要介绍的是使用 NCBI Primer-BLAST 网页工具来设计引物。
Primer-BLAST 是NCBI提供的一个在线工具,本质上是在他的服务器上运行 Primer3 来进行引物的设计,然后再利用NCBI丰富的各种序列数据库进行特异性比对,然后呈现出结果供我们筛选。
Step1:选择模板(要检测的基因mRNA或cDNA序列)
方法1:通过Nucleotide数据库找到目的基因的mRNA序列
在Primer Blast 工具里面的显示如下:
通常情况下,我们需要先找到我们目的基因的序列,打开NCBI或Pubmed的首页,找到 Nucleotide 数据库(Home – Nucleotide – NCBI (nih.gov))。
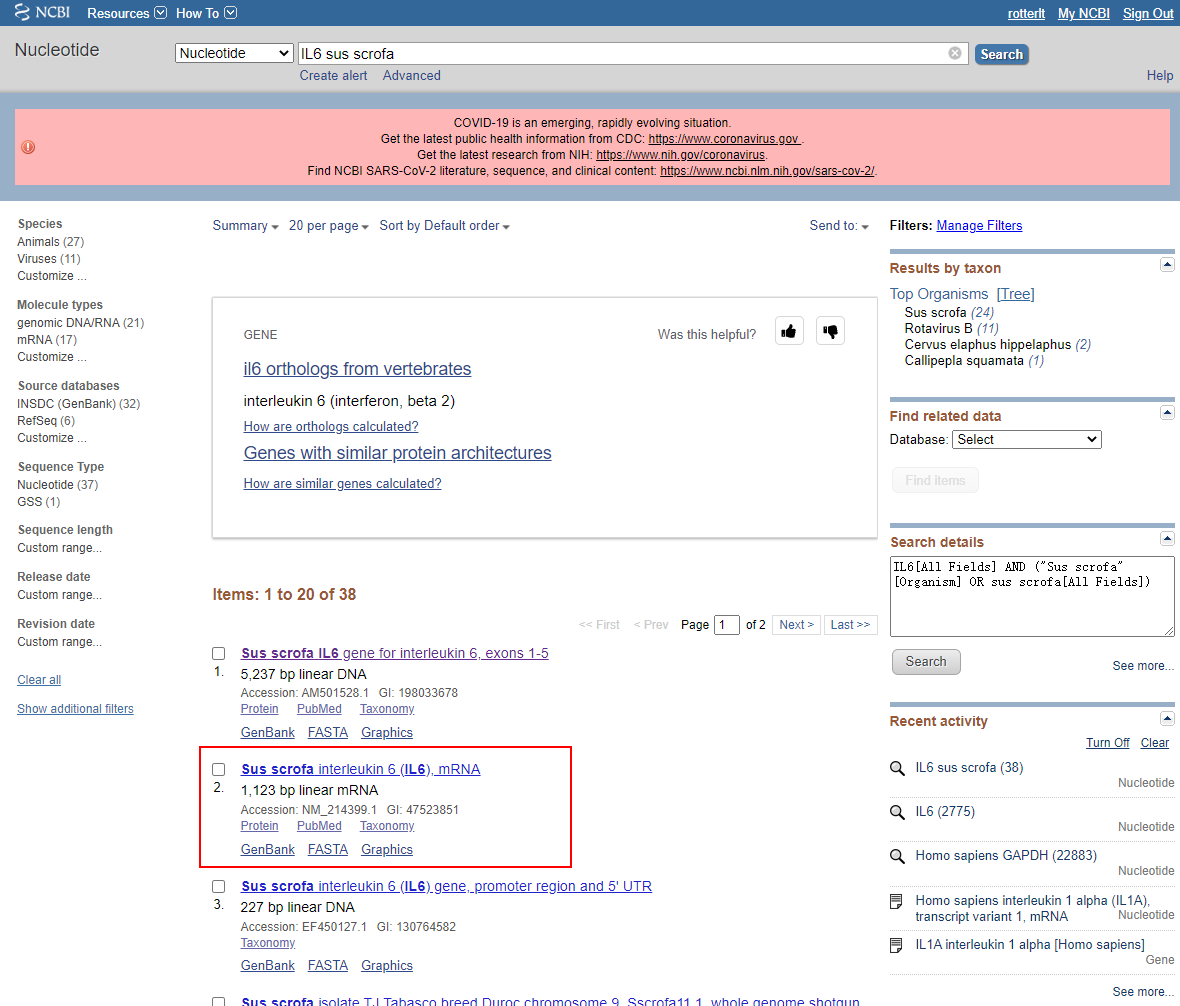
这里以猪(sus scrofa)的 IL6 为例,在搜索框中输入即可,可以同时输入物种名,也可以在右侧筛选Organisms:
我们选择mRNA的序列,也就是这里的第2条。
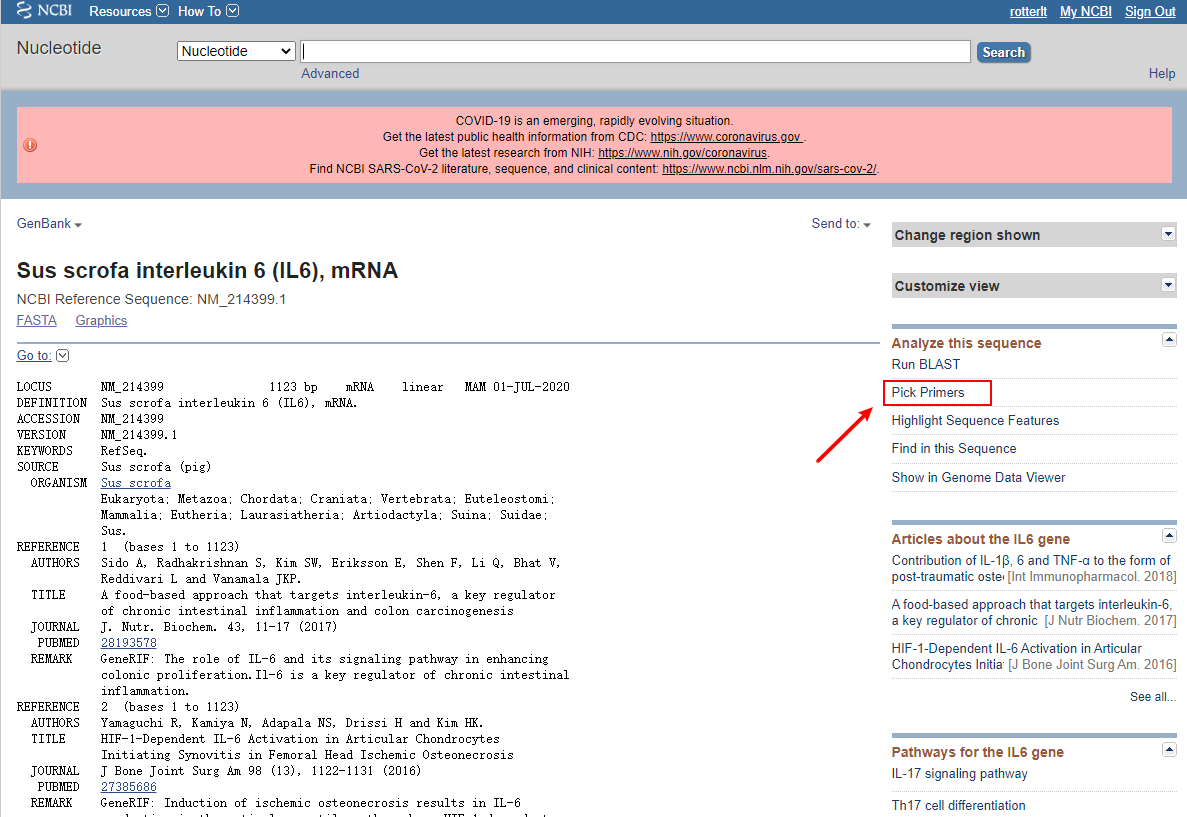
(也可以通过gene数据库先查到该基因的详细信息,再找到对应在Refseq mRNA中的序列信息。)
在Analyze this sequence 选项中找到Pick Primers,即可直接进入NCBI Primer-BLAST 界面,并且自动帮我们填好了序列信息和后面会涉及到的种属特异性的选择。
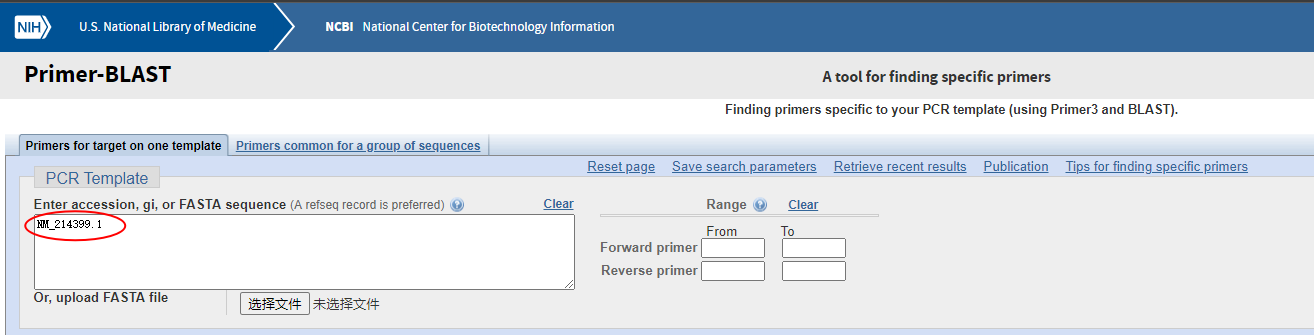
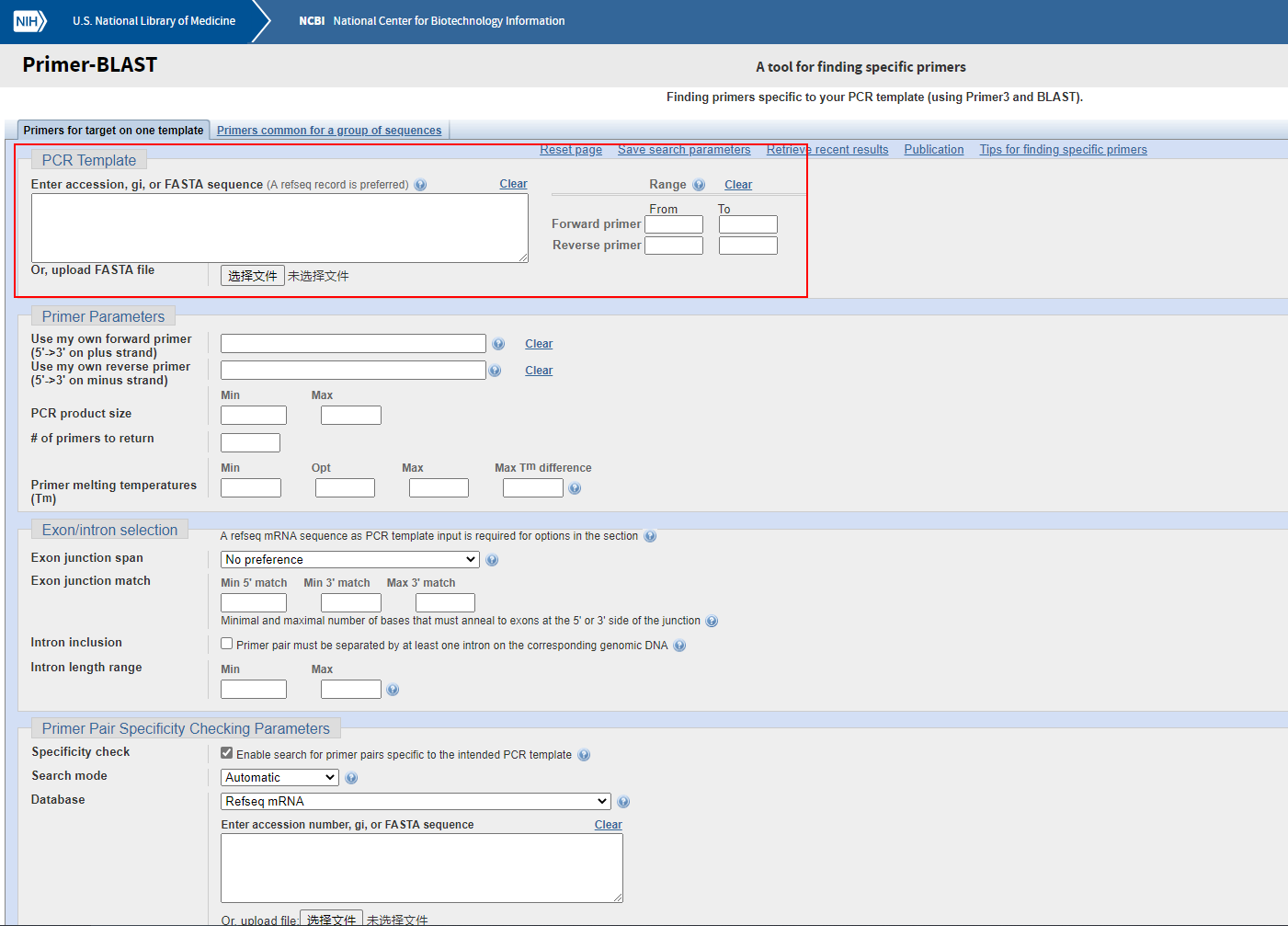
右侧“Range”选项中是可以选择我们要的前向引物和反向引物分别要在模板序列的什么位置,这里大多数情况下不需要指定。
方法2:已知序列的情况下
如果我们已知一个基因的accession或者gi编号,可以直接在这里输入进去,不需要查找序列即可开始设计。
如果我们有基因的序列文件,也可以上传FASTA文件。
Step2:引物基本参数设置
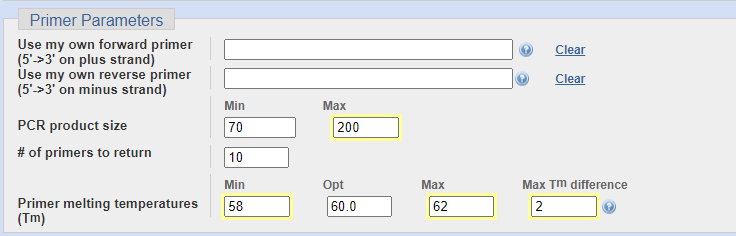
这一部分开头有两个是使用自己的序列选项,当我们使用别的工具设计的引物,我们要利用NCBI的数据库来检测引物特异性时填写,这里我们空着就行。
接下里可以填写产物的大小,一般情况下,50~300之间都问题不大,我通常使用70~200的范围。
# of primers to return 是说要服务器生成多少对引物来供我们参考选择,一般10个就够了,如果需要进行更加精细的比对和选择额,可以适当增加这个数值。
最佳Tm值选择60℃,范围可以选择58~62,前后引物的Tm差其实是越小越好,但是太小的话可能是找不到合适的引物,我一般选择2℃之内。
Step3:外显子/内含子设置
如果在Step1中提交的是FASTA文件,则无法进行外显子/内含子设置,因为只有在NCBI数据库内的序列,他才知道外显子/内含子都在哪。
必要知识
真核生物的基因是不连续表达的,转录过程中会首先生成hnRNA,然后通过一系列的剪切、剪接机制,去掉内含子,将外显子拼接在一起。
如果说我们设计的前后引物在同一个外显子上,那么无论模板是mRNA逆转录后(cDNA)还是gDNA(基因组DNA),它都可以扩增出来,如下图。
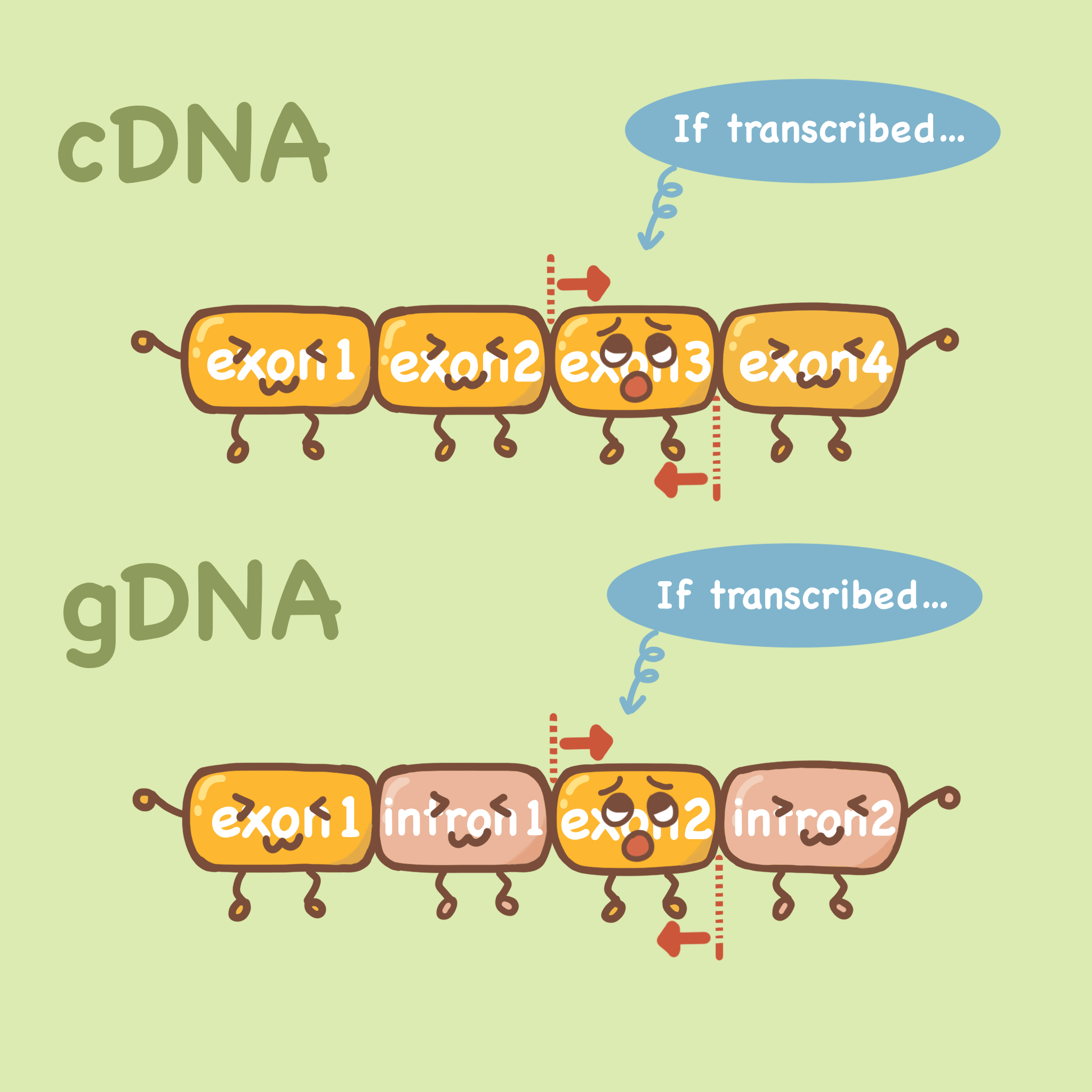
我们做qPCR检测某个基因的表达量,首先会提取RNA,如果在提取RNA的过程中没有完全去除基因组DNA,就会导致我们的逆转出来的cDNA实际上会含有gDNA污染,在qPCR的体系中,gDNA也会成为模板,最终导致我们的实验出现假阳性,实验结果不可靠。
所以我们这一步的设置,就是为了规避gDNA污染对实验结果带来的影响。
外显子设置
第一种方法是选择跨外显子连接的引物,如下图cDNA中,假如我们要扩增exon3,引物的序列是20bp左右,前向引物的配对跨越exon2和exon3,反向引物亦可跨外显子设计。那么在gDNA中,由于内含子的存在,跨外显子设计的引物无法牢固的结合,自然不能扩增出来。
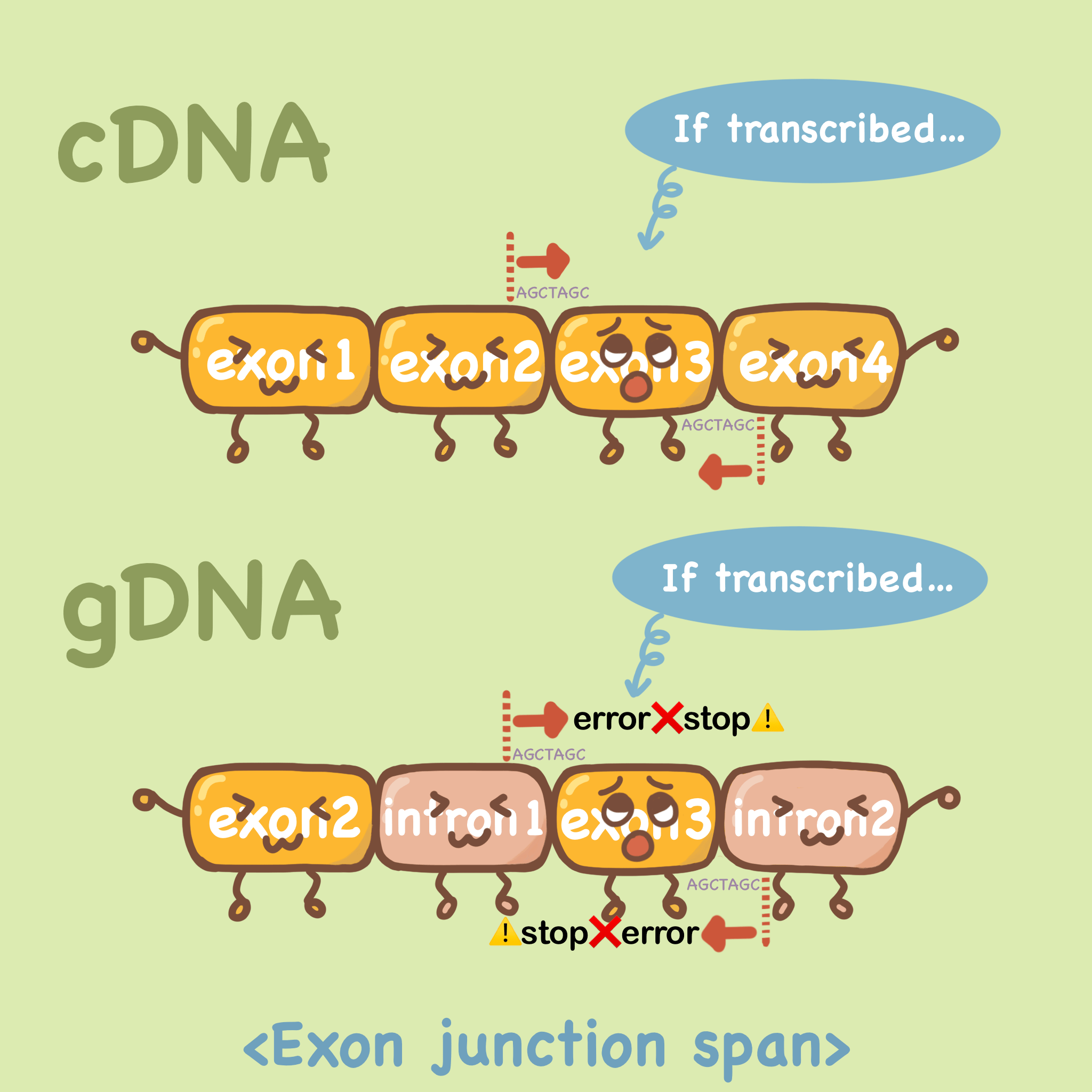
网页中Exon junction span选项中选择 “Primer must span an exon-exon junction” 。Exon junction match是选择结合部分的长度,保持默认即可。
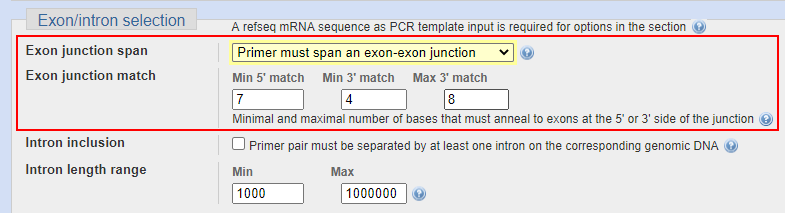
内含子设置
另一种方法是让前后引物分别在两个外显子上,中间横跨一个内含子。假设模板中有gDNA污染,那么我们可以扩增出来一长一短两个产物,长的这个就是以gDNA为模板扩增出来的。
由于内含子的序列有长有短,为了确保gDNA不影响实验结果,我们通常将跨过的内含子长度最小值设置在1000,事实上,超过500bp的产物在qPCR的体系中基本上就很难扩增出来了。
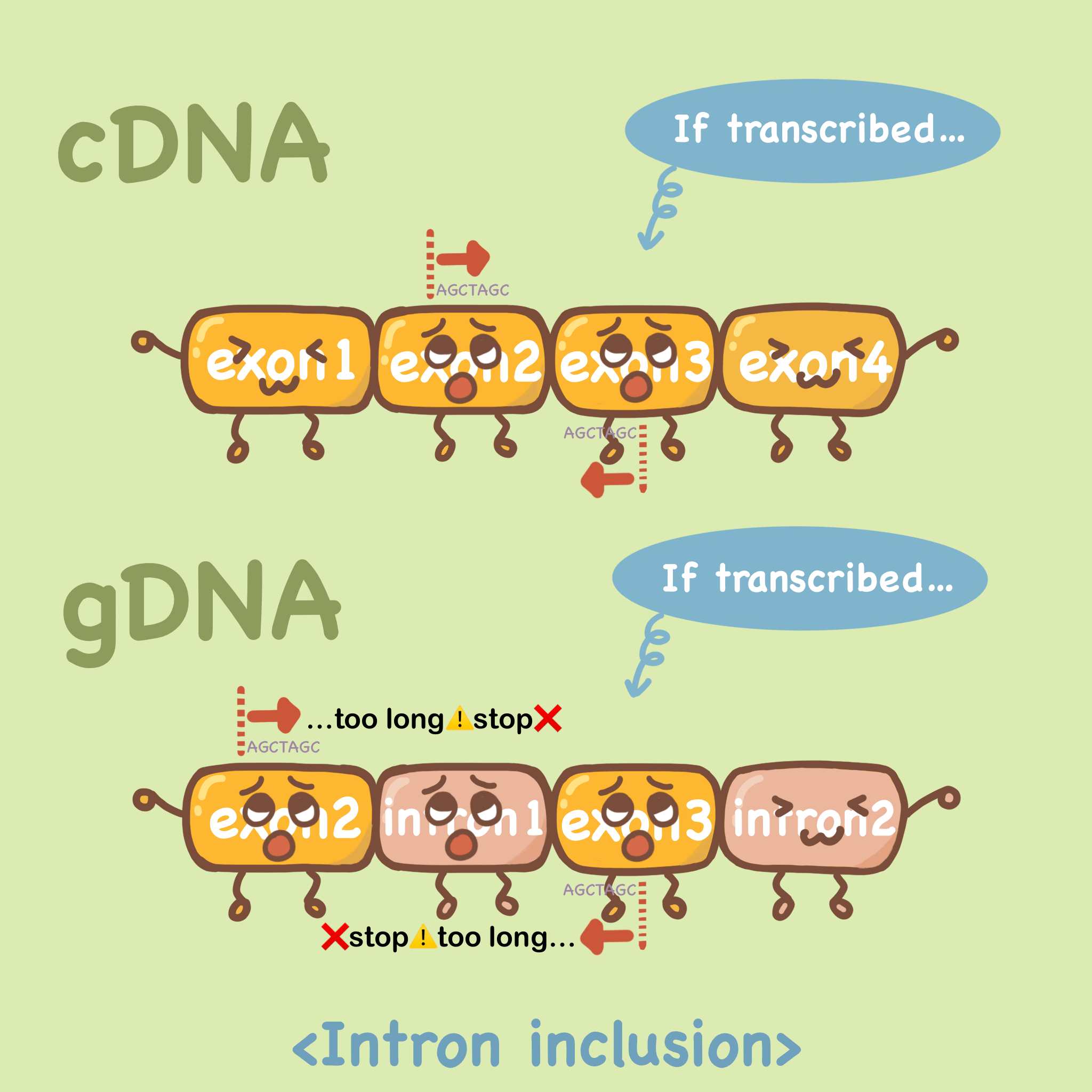
通过这种设置,即使模板中有gDNA污染,它也无法进行扩增,对结果也就没有影响。
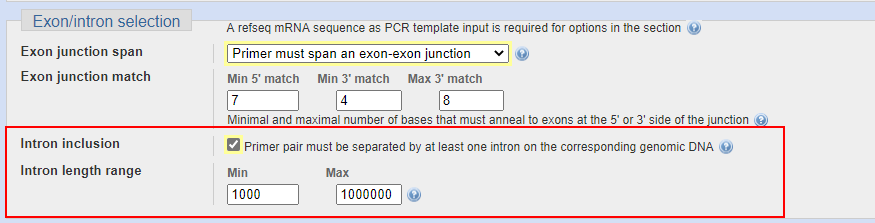
此项设置只需要勾选即可打开,并且它可以和外显子同时设置,也可以单独只选这个。
我自己的习惯是 只设置横跨内含子,而不要求跨外显子连接,这样保证引物质量的同时也可以找到更多的引物供选择。如果要求过于严格很容易出现找不到合适的引物的情况。
Step4:设置引物特异性检查
引物的特异性指的是这对引物在我们的体系中是否只能扩增出我们要的目的基因,而不会有其他的乱七八杂的产物。一般我们的样品中只有某个物种的cDNA,假设是猪sus scrofa (taxonomy id=9823),那我们进行引物特异性检查的时候就只需要比对猪的mRNA参考序列的数据库即可,而不用检查所有的物种。
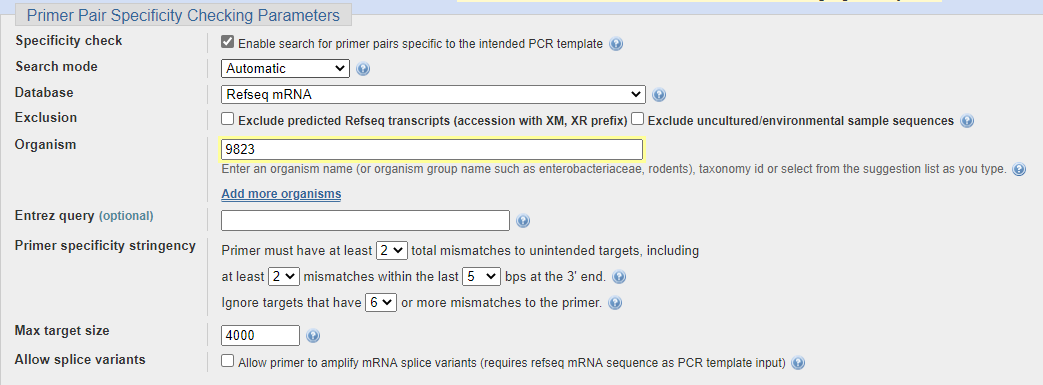
通常情况下,这一系列选项中只要注意一下 Organism,这里要输入物种的拉丁文名称或者taxonomy id 即可。在第一步选择模板序列时如果是在nucleotide数据库中点击 pick primers 跳转过来的,系统会自动选择相应的物种,否则需要自己填写。其余选项保持默认即可。
对于一些特殊的情况,如果需要在多个物种中进行特异性检查,可以在下方点击 Add more organisms 就可以增加。
在Get Primers 按钮下方,还有一个高级选项的下拉菜单,里面还有丰富的可以修改的参数,比如说产物的Tm值等,这些内容不修改也没问题,具体怎么用大家自行探索吧。
Step5:选择合适的引物对
点击底部的Get Primers按钮,可以勾选在新窗口打开结果,这样如果出来的结果不是很满意或者可供选择的结果很少,可以方便修改参数再次生成。

这个例子是生成的一个猪的 IL6 的 qPCR 引物,可以看到Primer-BLAST给出的结果非常直观,会明确列出引物序列、长度、Tm值、自身配对等参数,除此以外,由于我要求了跨内含子设计引物,所以这里有一个 Total intron size ,告诉我们这对引物中间跨过的内含子长度。
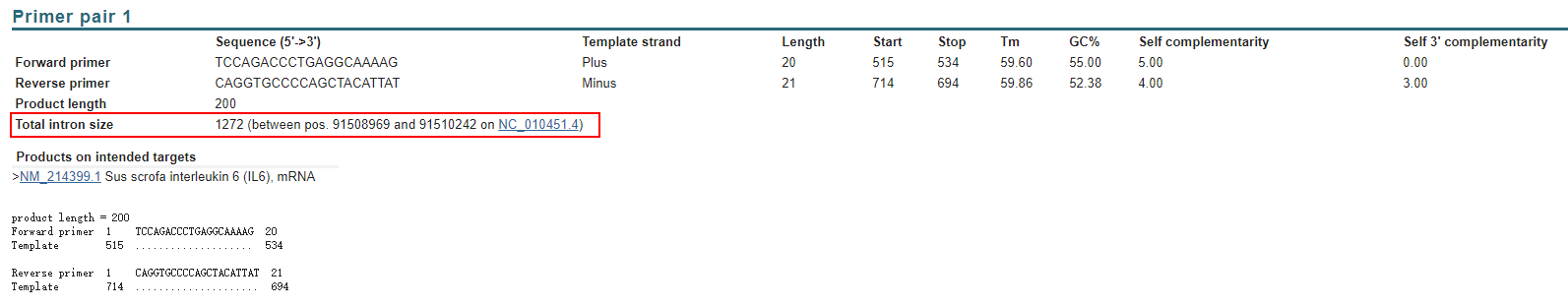
这样引物就设计完成啦!可以愉快的进行后续实验了!
References
- NCBI/Primer‐Blast: Finding primers specific to your PCR template (using Primer3 and BLAST). Available at: http://www.ncbi.nlm.nih.gov/tools/primer‐blast/ .
- Thornton B, Basu C. Real-time PCR (qPCR) primer design using free online software. Biochem Mol Biol Educ. 2011 Mar-Apr;39(2):145-54. doi: 10.1002/bmb.20461. PMID: 21445907.
- 更严谨的 qPCR引物设计方案(SYBR Green).https://mp.weixin.qq.com/s/qY2N3l1wye72jmxmYmixMA
- 生工技术 | [万字长文] Primer-BLAST设计qPCR引物. https://mp.weixin.qq.com/s/k-bRxEkJLe565Ry9AD2DuA



暂无评论内容