
scRNA-seq共19篇
排序
scRNA-seq数据整合Introduction
Seurat不仅可以校正实验的批次效应,还能跨平台整合数据,例如将10x单细胞数据、BD单细胞数据和SMART单细胞数据整合在一起;也能整合单细胞多组学数据,例如将单细胞ATAC、空间转录组与单细胞转...
归一化与主成分分析-HBC lesson 5
在我们我们获得高质量的单细胞数据后,单细胞RNA测序分析工作流程的下一步是进行聚类。聚类的目标是将不同的细胞类型分离成独特的细胞群。为了执行聚类,我们需要确定细胞间表达差异最大的基因...
cellranger的安装
新建一个文件夹 打开linux终端,运行 mkdir yard 验证一下工作目录 pwd /home/vamond/yard 进入该目录,新建apps文件夹,用于存放cellreanger cd /mnt/home/user.name/yard mkdir apps cd apps ...
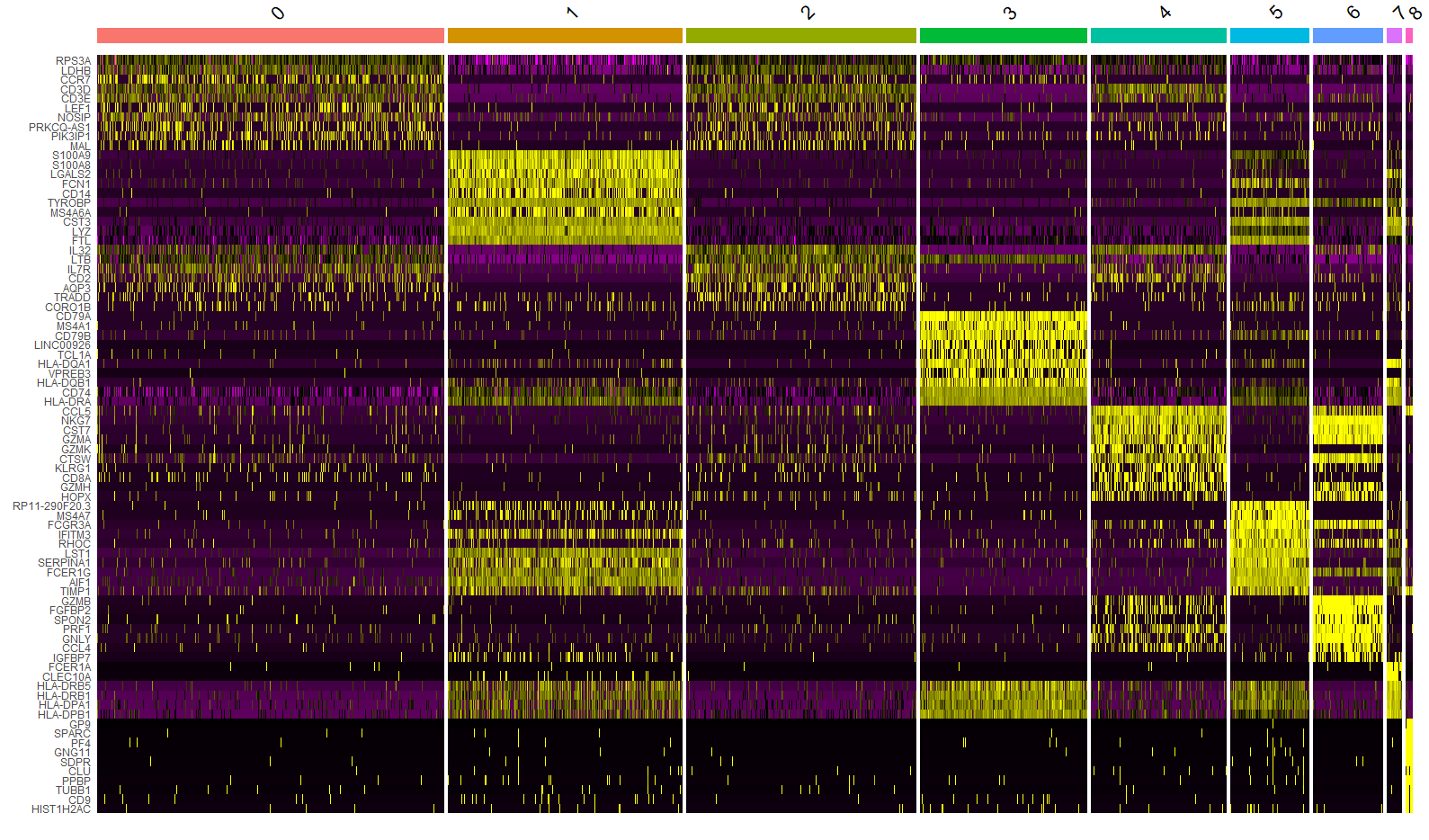
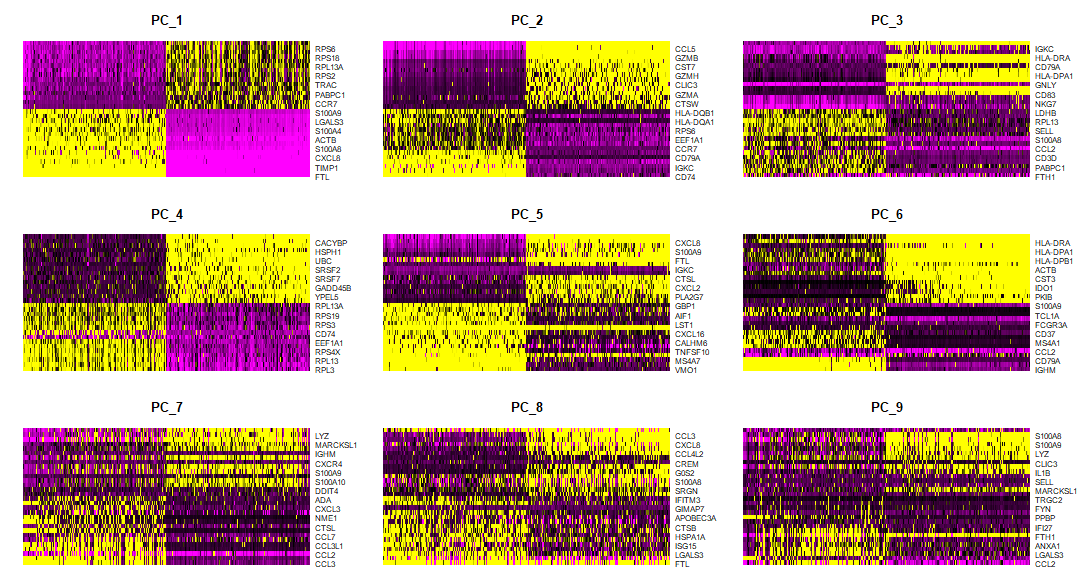
使用pheatmap可视化marker基因
在完成单细胞分析基本流程之后,我们获得了各个细胞聚类和相应的marker基因,有多种方式可以可视化marker基因的表达量,seurat包中自带的DoHeatmap()、VlnPlot()以及DotPlot()函数可以很方便的...
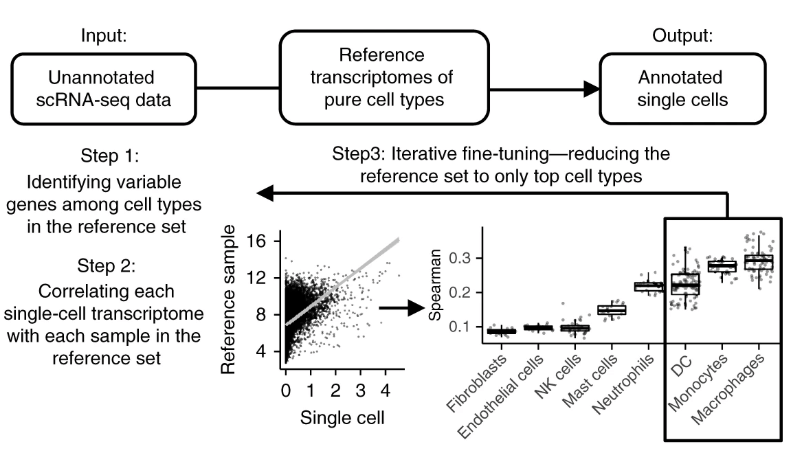
使用singleR预测细胞类型
SingleR是用于单细胞RNA测序(scRNAseq)数据的自动注释方法(Aran et al.2019)。给定具有已知标签的样本(单细胞或RNAseq)参考数据集,它将基于与参考数据的相似性标记测试数据集中的新细胞...
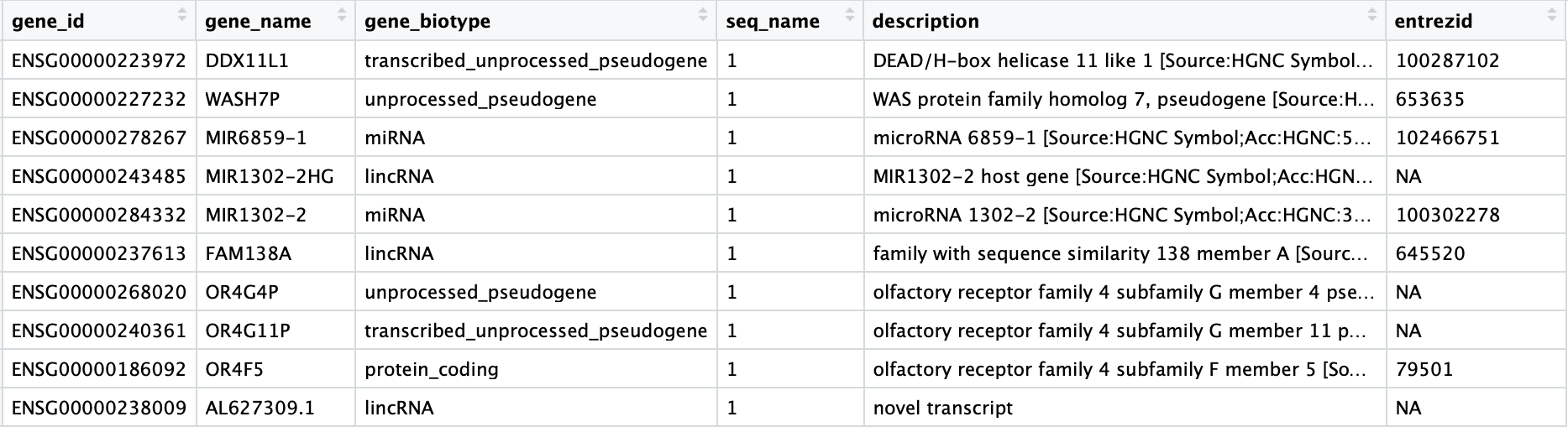
通过注释文件计算线粒体序列比例
经验证,人类样本的单细胞分析使用seurat教程的方法计算线粒体序列比例与此方法结果一致,但对于其他物种,建议使用此方法。 使用注释文件生成线粒体计数指标 我们将使用AnnotationHub,它允许...
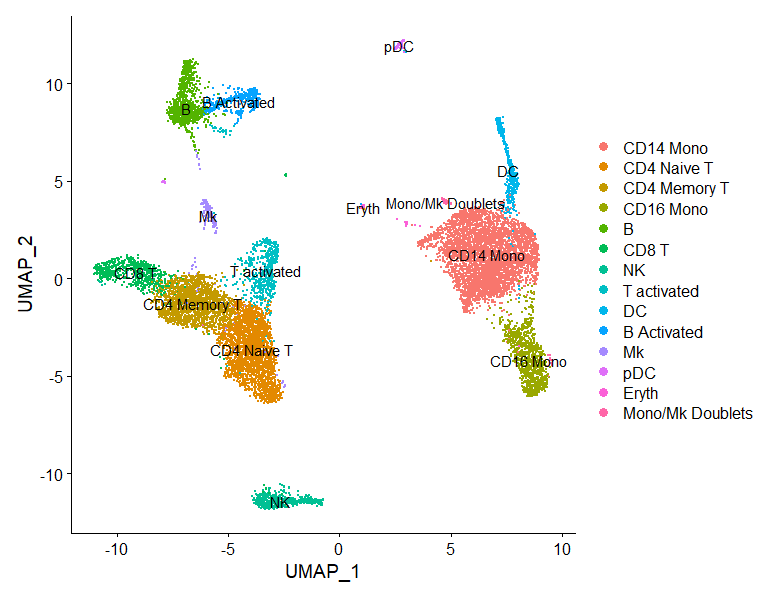
整合后的细胞标记物鉴别-HBC lesson 9
目标: 确定每个类群的基因标记物 使用标记物识别每个类群的细胞类型 根据细胞类型标记物来判断是否需要重新分组,或许需要合并或拆分聚类的类群 挑战: 对结果的过度解读 结合不同类型的标记物...

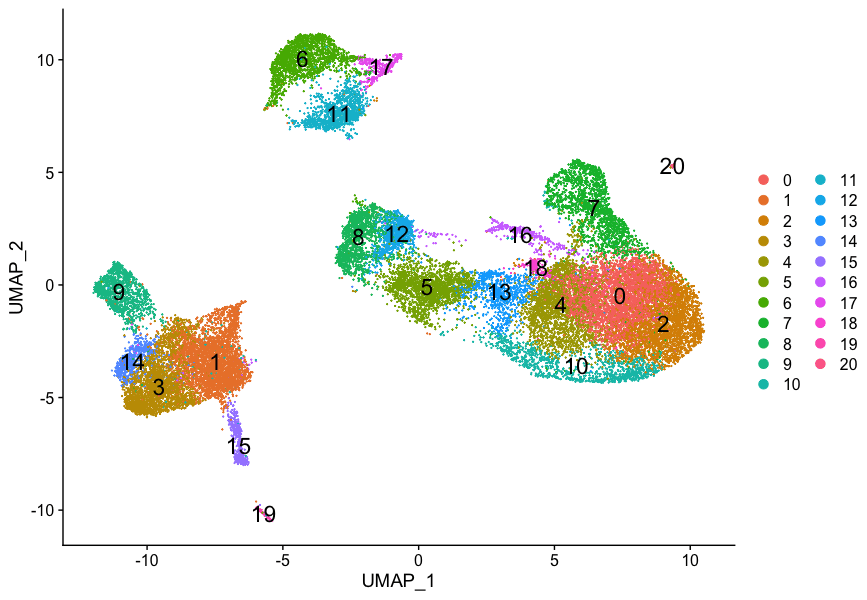
细胞聚类的质控-HBC lesson 8
学习内容 评估是否存在聚类的假象 用PCA和UMAP图来确定聚类的质量,并了解何时需要重新聚类 评估已知的细胞类型标志物以假设集群的细胞类型身份 目标 确定集群是否代表真正的细胞类型或由于生物...
聚类分析-HBC lesson 7
学习内容 学会选择合适的PCs用于聚类分析 聚类的方法 目标 产生细胞类型特异性聚类,并使用已知的细胞类型标记基因来确定聚类的身份。 确定集群是否代表真正的细胞类型或由于生物或技术差异而产...
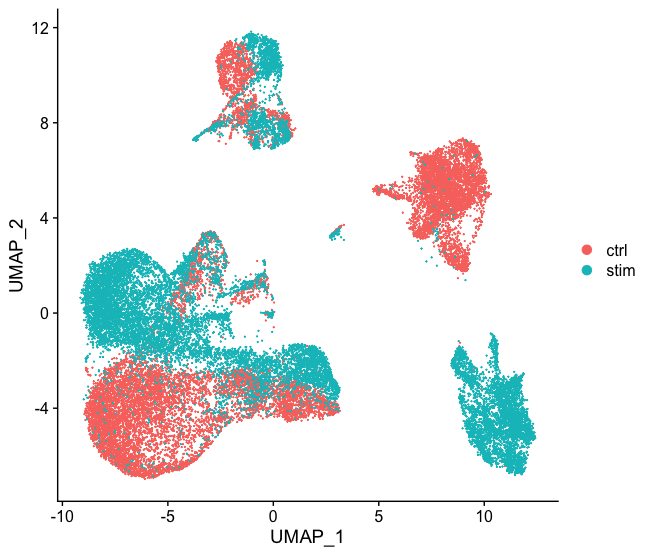
整合-HBC lesson 6.2
本文主要是翻译,用作参考,还是要看GitHub上的英文原版学习 目标 对于不同条件下的样本,将相同类型的细胞对齐到一起 挑战 对准类似的细胞类型,这样我们就不会因为样品、条件、模式或批次的不...