
生信共42篇
生物信息学教程和笔记

.rds和.Rdata(.rda)
.rds和.Rdata (也称为.rda )文件都可用于以R本机格式存储R对象。与非本机存储方法(例如write.table相比,保存此方法有多个优点: 1)将数据恢复到R更快 2)它保持在数据中编码的R...

linux常用命令:tar
Linux tar(英文全拼:tape archive )命令用于备份文件。 tar是用来建立,还原备份文件的工具程序,它可以加入,解开备份文件内的文件。 备份文件不完全等同于压缩文件,linux中的备...
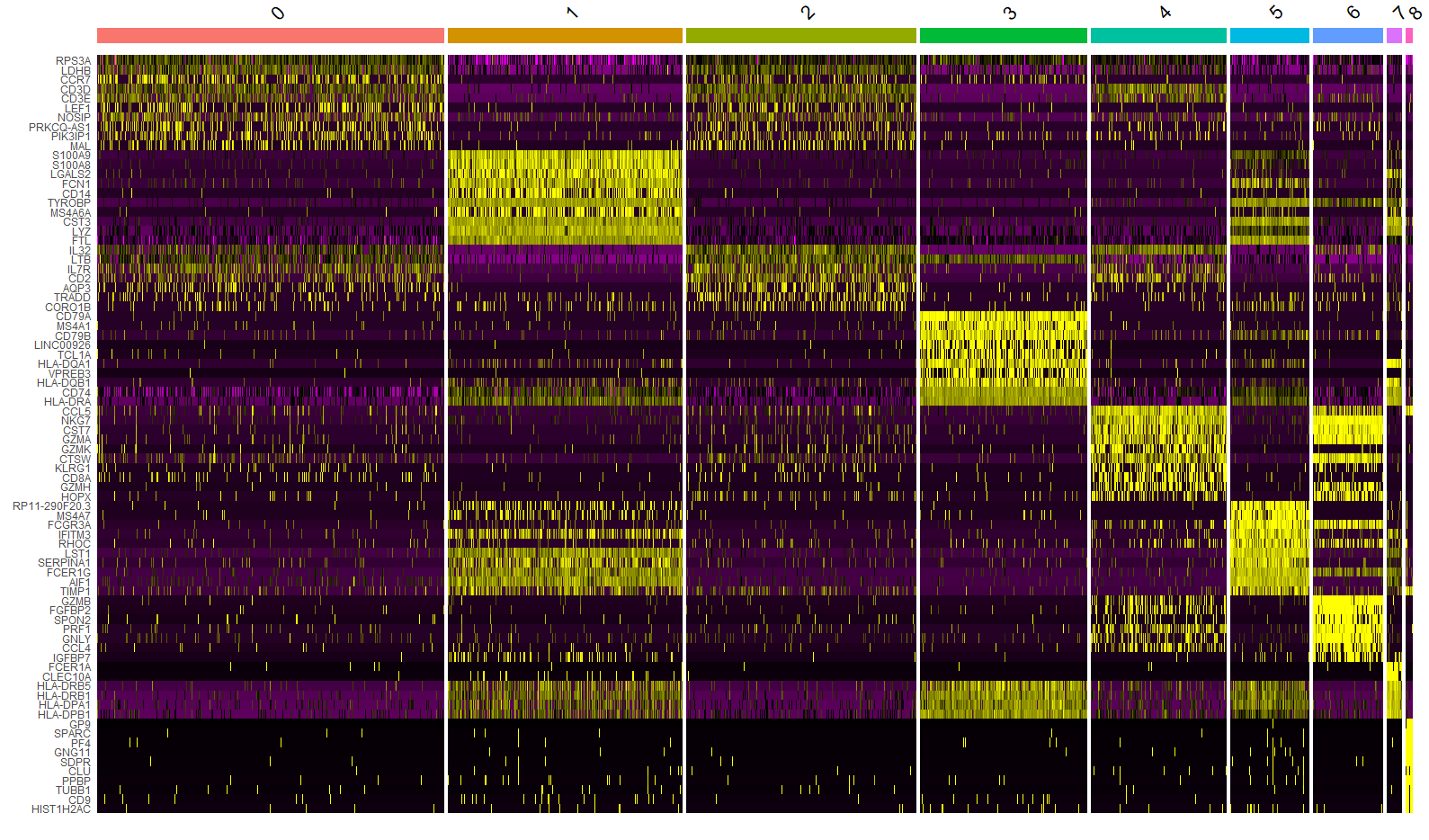
使用pheatmap可视化marker基因
在完成单细胞分析基本流程之后,我们获得了各个细胞聚类和相应的marker基因,有多种方式可以可视化marker基因的表达量,seurat包中自带的DoHeatmap()、VlnPlot()以及DotPlot()函数可以很方便的...
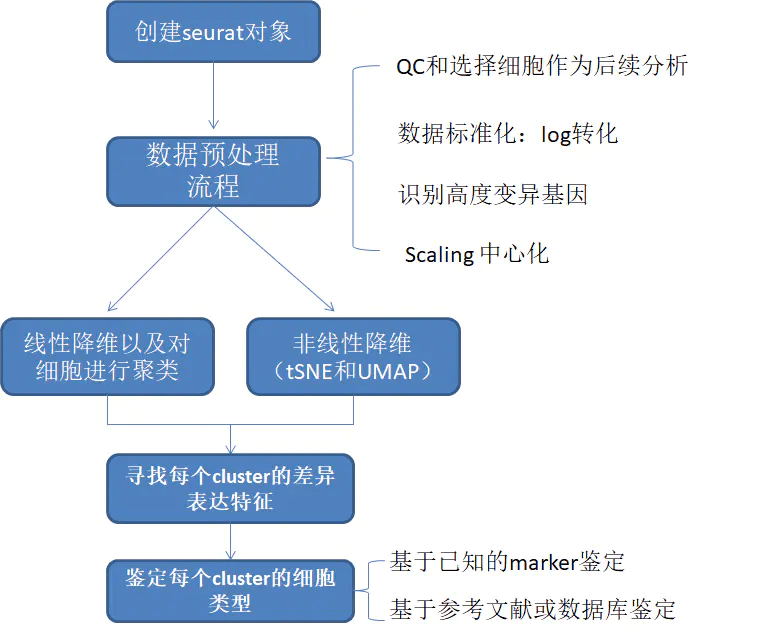
Seurat基本分析流程
参考链接: https://satijalab.org/seurat/articles/pbmc3k_tutorial.html 建立 Seurat 对象 示例数据为10X Genomics的外周血单个核细胞(PBMC)数据集,含有2700个单细胞,使用Illumina NextSeq...
使用Aspera高速下载ngs数据并进行MD5校验
因为课题研究需要结合参考一篇已发表的单细胞数据,由于该文章只提供的原始fastq文件,所有要下载下来从上游分析开始做起。(如果是是做数据库生信挖掘的也需要下载文件哦)一开始尝试的使用htt...
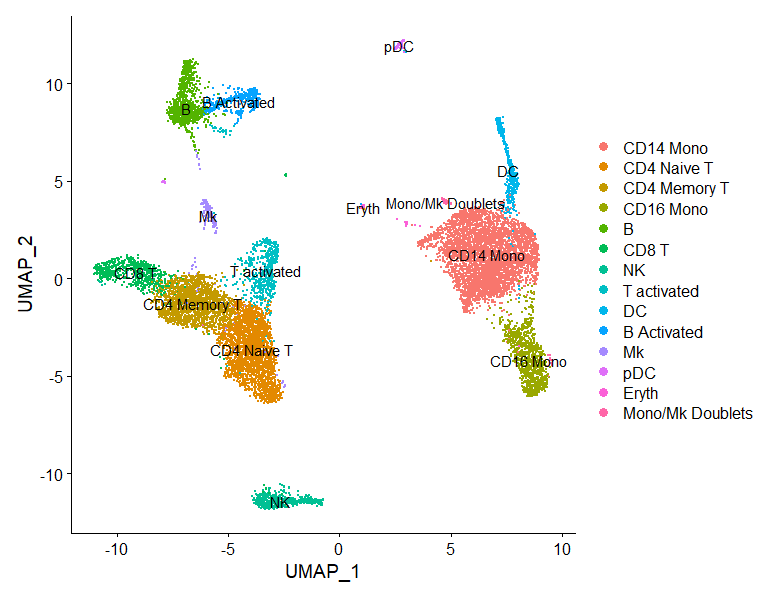
scRNA-seq数据整合Introduction
Seurat不仅可以校正实验的批次效应,还能跨平台整合数据,例如将10x单细胞数据、BD单细胞数据和SMART单细胞数据整合在一起;也能整合单细胞多组学数据,例如将单细胞ATAC、空间转录组与单细胞转...
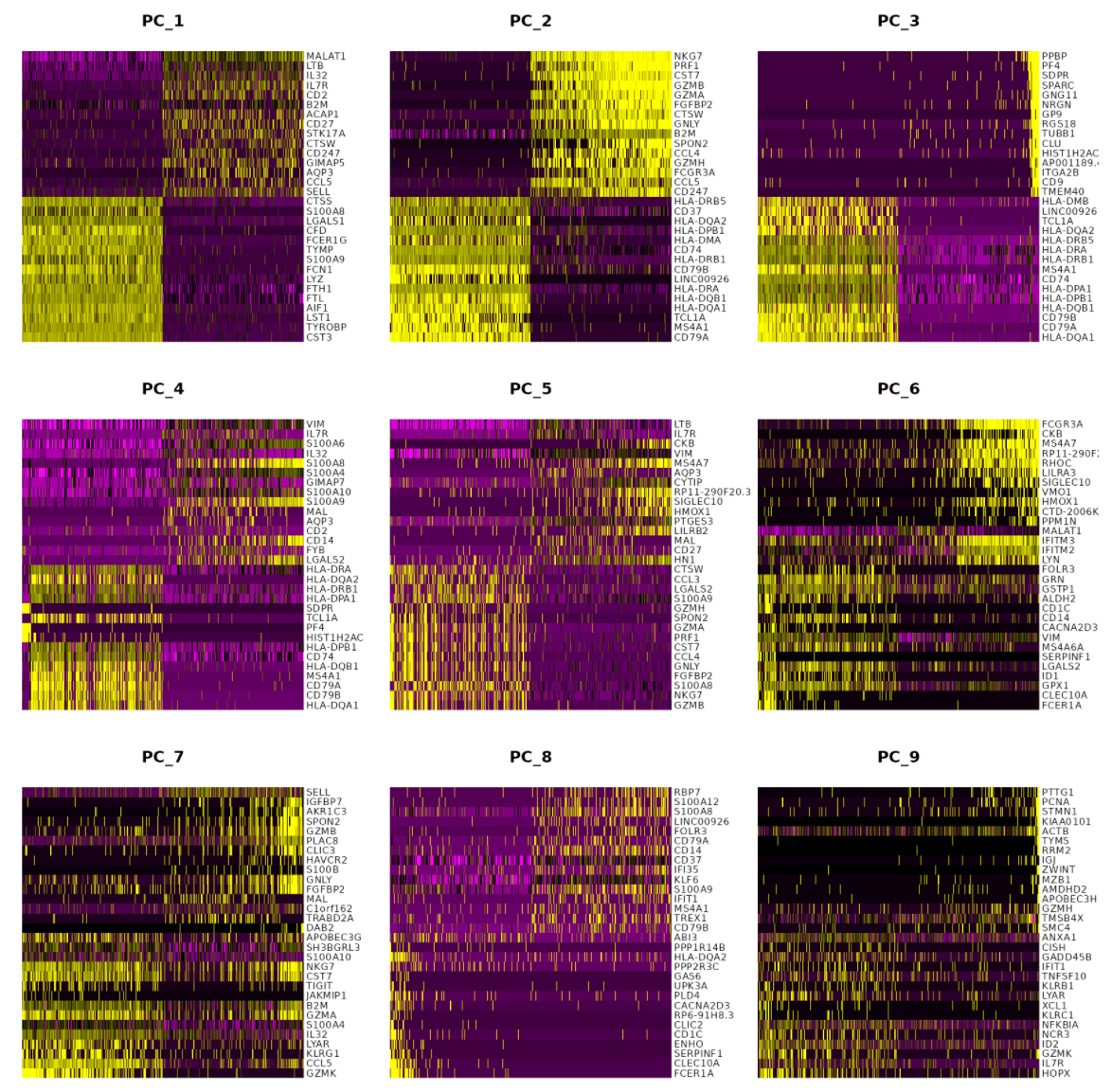
关于“数据的维度”(dims参数)的选择
关于“数据的维度”(dims参数)的选择 Created time: Apr 13, 2021 12:07 PM Tags: R, Seurat, scRNA-seq 完成PCA之后,我们获得了该数据集的所有主成分(PCs)信息,但是如何决定纳入多少个主成...
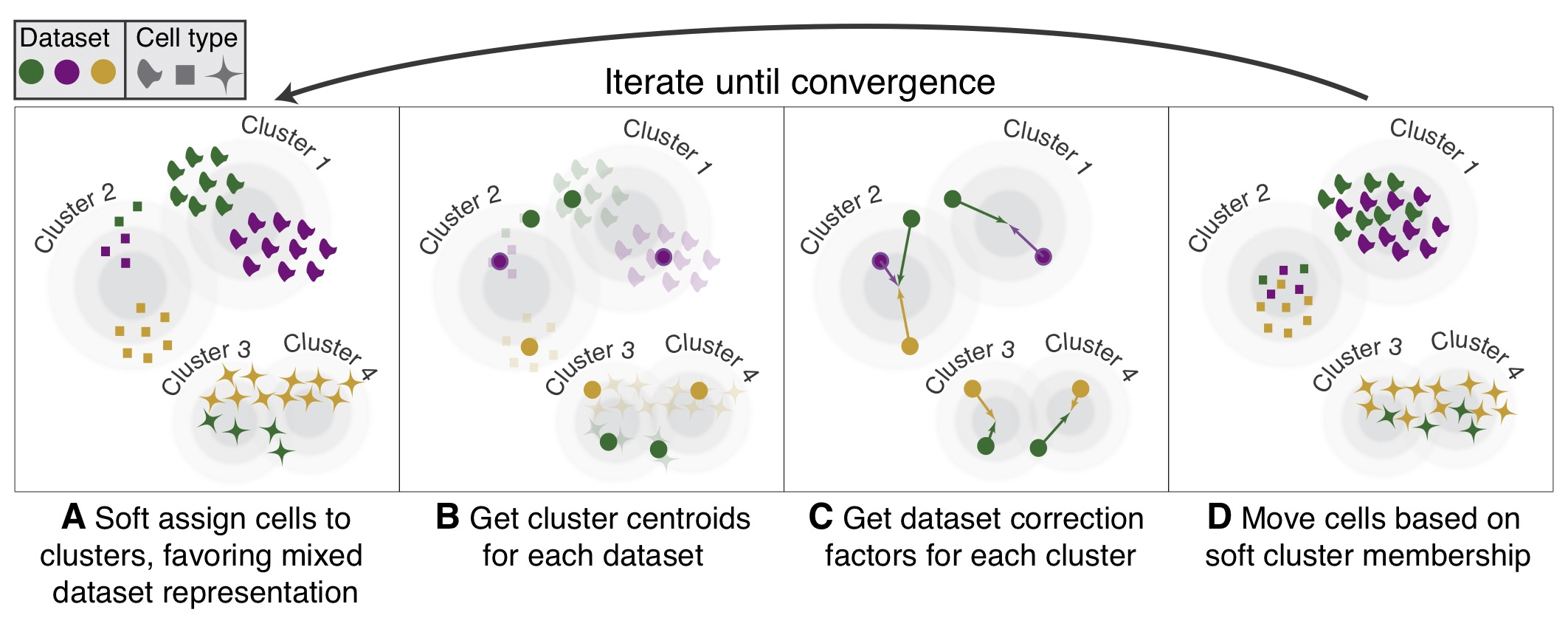
对Seurat对象使用harmony方法整合
Harmony方法在2019年发表在上面nature methods, harmony算法与其他整合算法相比的优势: 整合数据的同时对稀有细胞的敏感性依然很好; 省内存; 适合于更复杂的单细胞分析实验设计,可以比较来...

cellranger count 流程
准备工作:下载参考基因组和数据集 下载参考基因组 # 新建一个ref文件夹存放参考基因组 cd yard mkdir ref cd ref # 下载human GRCH38,大约11G wget https://cf.10xgenomics.com/supp/cell-exp...
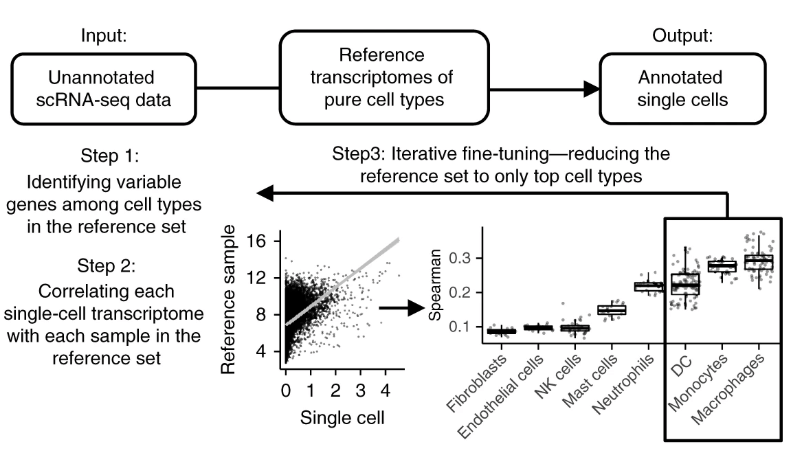
使用singleR预测细胞类型
SingleR是用于单细胞RNA测序(scRNAseq)数据的自动注释方法(Aran et al.2019)。给定具有已知标签的样本(单细胞或RNAseq)参考数据集,它将基于与参考数据的相似性标记测试数据集中的新细胞...