
单细胞 第3页
学习单细胞分析过程中的一些笔记,多数来自网络已有教程,我记录了一下自己的学习过程,并且整理在此,以供后续参考。

scRNA-seq简介-HBC lesson 1
参考链接: https://github.com/hbctraining/scRNA-seq_online/blob/master/lessons/01_intro_to_scRNA-seq.md 我们为什么需要单细胞RNA测序 人类各种组织之间细胞的类型,状态和相互作用差异巨...

“归一化”与“标准化”
参考链接: https://mp.weixin.qq.com/s/6ioR3JE0wKg6M-YAsLBcTA 关于归一化和标准化的翻译看了很多中文资料,发现还是有争议的,在seurat中主要是两个函数:NormalizeData()和ScaleData() ,其...

单细胞聚类resolution参数的选择
在进行细胞聚类时有两个参数的选择对下游分析的结果影响很大 一个是纳入分析的PCs数,也就是dims参数,选择方法见 关于“数据的维度”(dims参数)的选择 ; 另一个是resolution参数,官方推荐根...
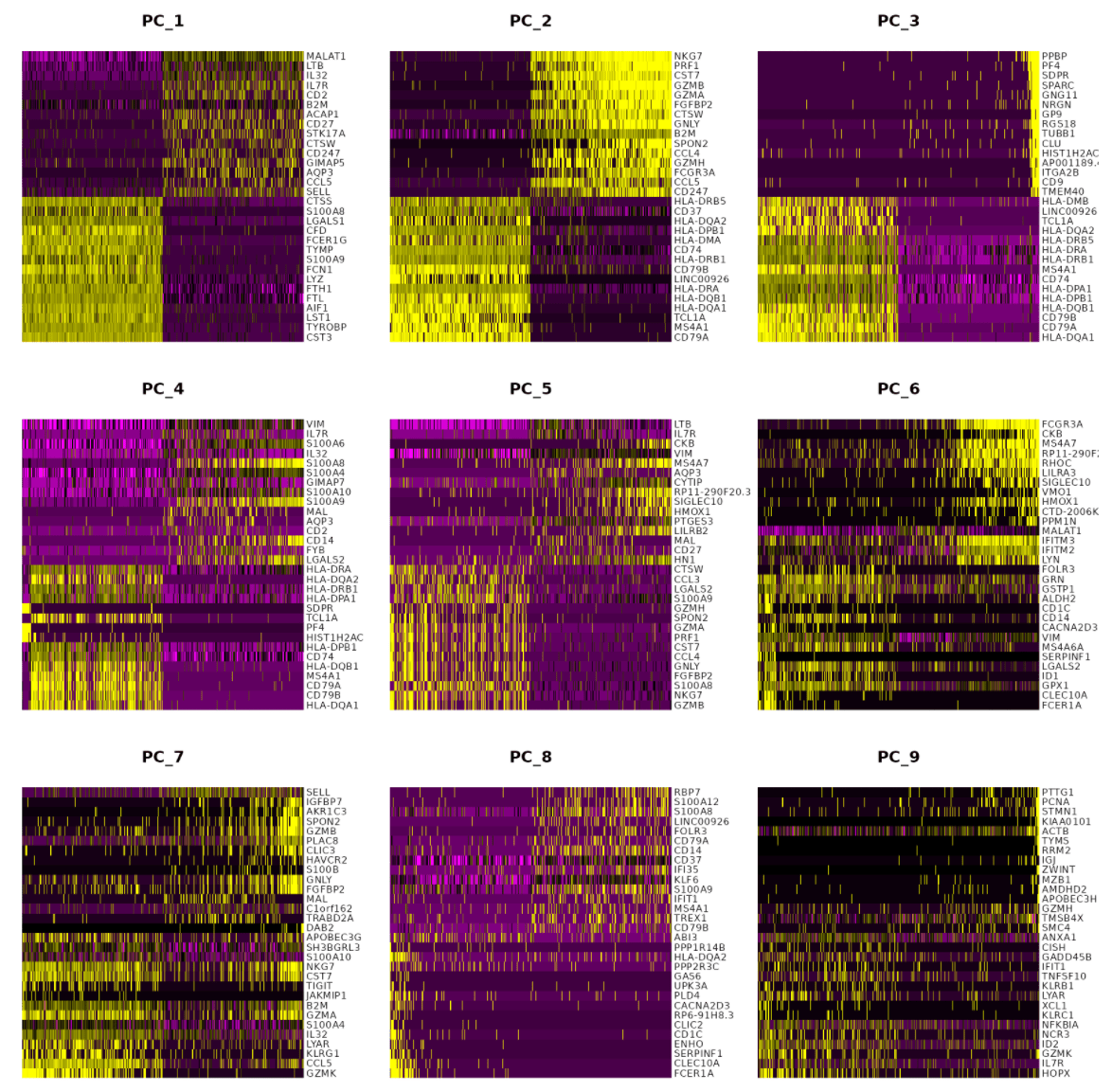
关于“数据的维度”(dims参数)的选择
关于“数据的维度”(dims参数)的选择 Created time: Apr 13, 2021 12:07 PM Tags: R, Seurat, scRNA-seq 完成PCA之后,我们获得了该数据集的所有主成分(PCs)信息,但是如何决定纳入多少个主成...
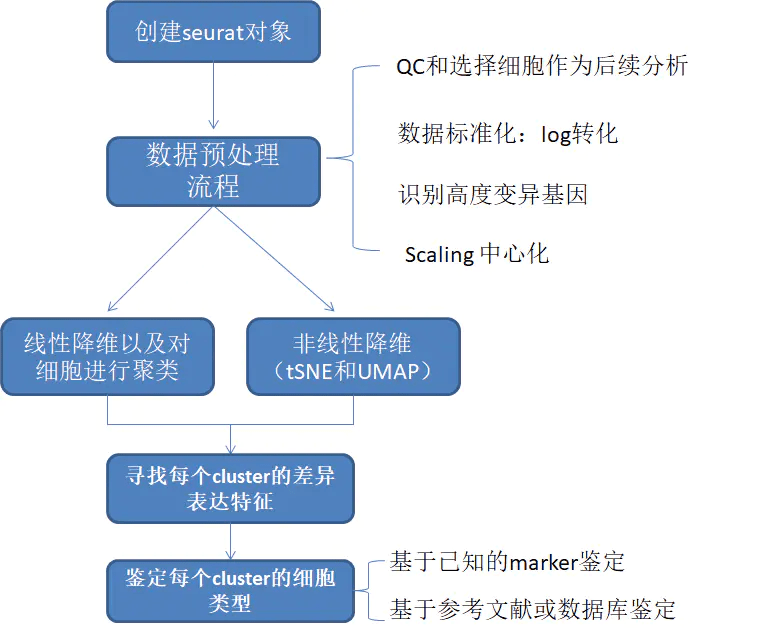
Seurat基本分析流程
参考链接: https://satijalab.org/seurat/articles/pbmc3k_tutorial.html 建立 Seurat 对象 示例数据为10X Genomics的外周血单个核细胞(PBMC)数据集,含有2700个单细胞,使用Illumina NextSeq...